|
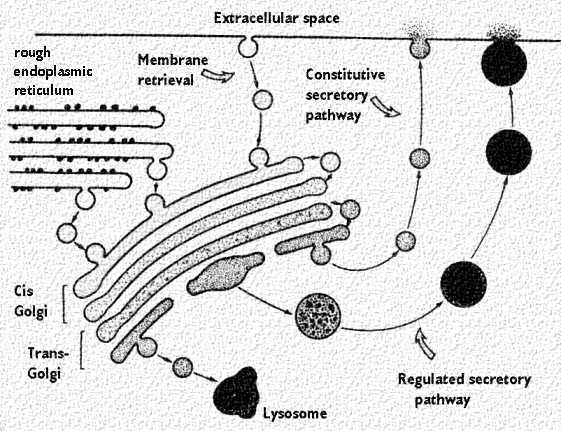
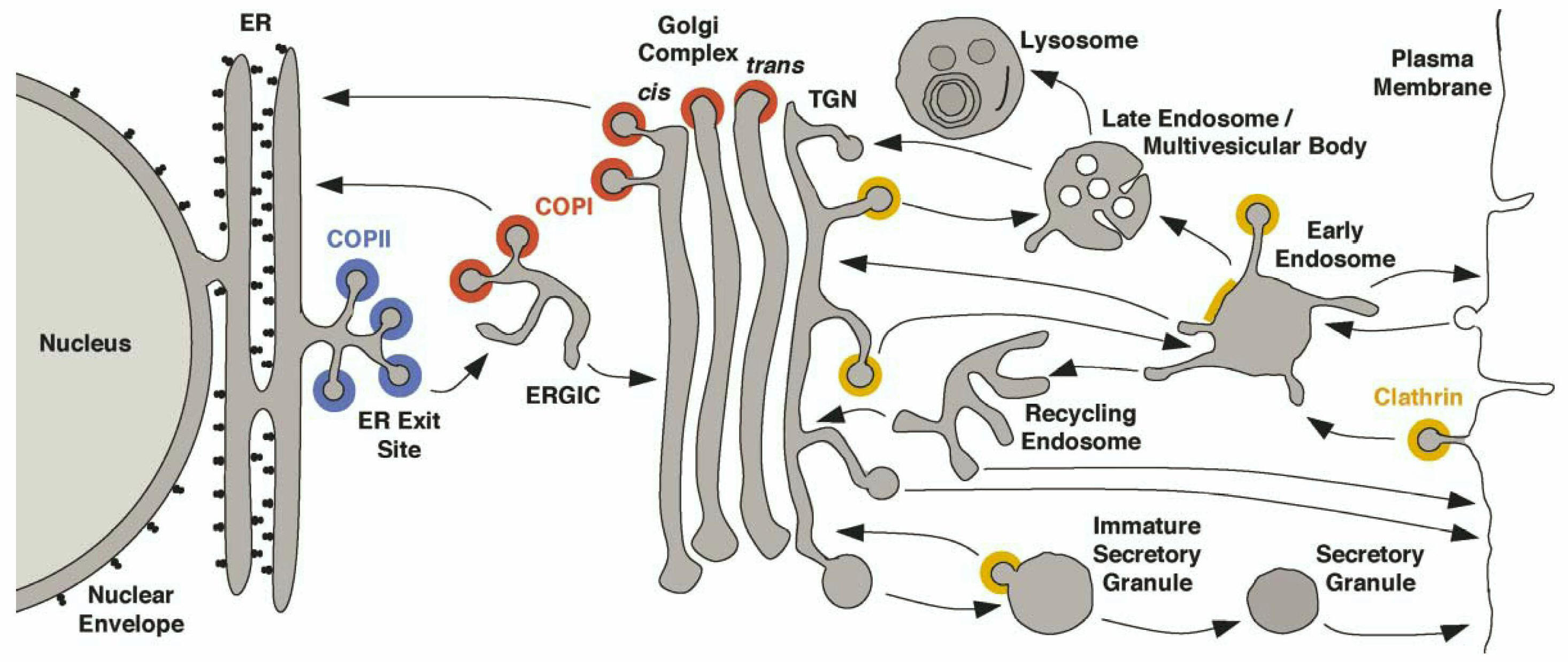
The Golgi complex
|
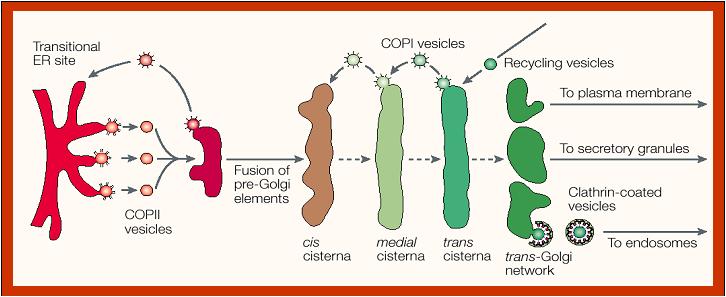
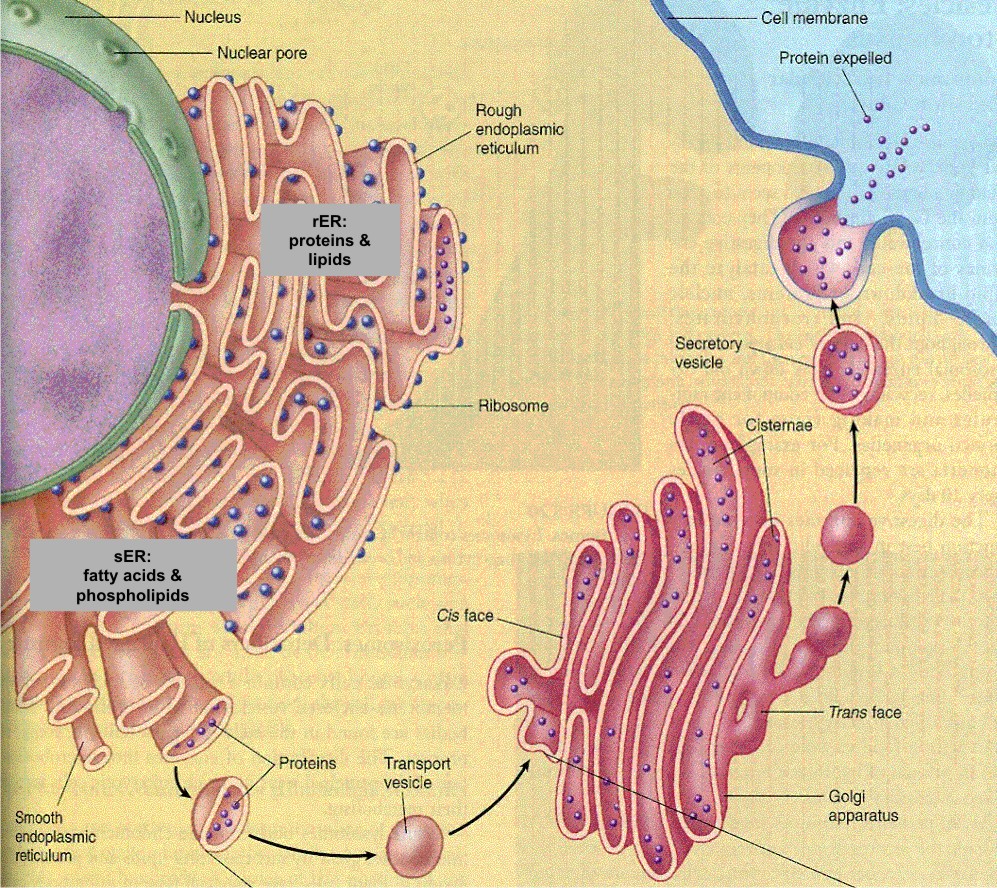
Once
secretory proteins are properly folded and modified in the first
compartment of the secretory pathway, the ER,
the proteins move to specialized regions, the ER export sites, and are packaged
in coated, irregularly shaped transport vesicles.
Following uncoating, the transport
vesicles form vesicular-tubular clusters (VTCs, also called the ER-Golgi
intermediate compartment ERGIC) that move to the Golgi.
The scheme below
depicts the compartments of the secretory pathway and two compartments
associated with the secretory pathway, namely the lysosomal/vacuolar and
endocytic pathways. Transport steps are indicated by arrows. Colors
indicate the known or presumed locations of the transport vesicle coats COPII (blue), COPI (red),
and clathrin (yellow/orange). Clathrin coats are heterogeneous and contain
different adaptor and accessory proteins at different membranes. Only
the function of COPII in ER export and of plasma membrane-associated
clathrin in endocytosis are known with certainty. Less well understood
are the exact functions of COPI at the ERGIC and Golgi complex and of
clathrin at the TGN, early endosomes, and immature secretory granules.
The pathway of transport through the Golgi stack is still being
investigated, but is generally believed to involve a combination of COPI-mediated
vesicular transport and cisternal maturation (see below). Additional coats or
coat-like complexes exist but are not represented in this figure. |
|
The
Golgi
apparatus
modifies
and
sorts
proteins
for
transport
throughout
the
cell.The
Golgi
apparatus
is
often
found
in
close
proximity
to
the
ER
in
cells.
Protein
cargo
moves
from
the
ER
to
the
Golgi,
is
modified
within
the
Golgi,
and
is
then
sent
to
various
destinations
in
the
cell,
including
the
lysosomes
and
the
cell
surface.
|
|
|
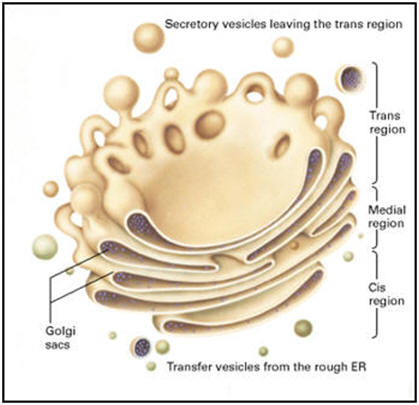
The
buds emerging from the ER thus become vesicles and are coated with COPII
protein coats. After the vesicles lose their COPII coat, they merge with
the VTCs (ERGIC) carrying the soluble and membrane proteins to the Golgi
complex. Note that the vesicles are moving to contribute to the
cis-Golgi network of vesicles and cisternae. The movement of these
special transport vesicles is an energy-requiring process. If one blocks
production of ATP, the transport will not happen. The drawing above shows how
the ER forms vesicles (without ribosomes attached) that carry the newly
synthesized proteins to the Golgi complex. The inside of the vesicle
becomes continuous with the inside of the Golgi cisternae, so that
protein groups pointing towards the inside, could eventually be directed
to face the outside of the cell. Carbohydrate groups are attached and
any subunits may be joined in these cisternae. The protein is then
passed to the final region of the Golgi called the "trans face". There
it is placed in vacuoles that bud from this region of the Golgi complex.
These may be a certain size or density, characteristic of the cell
itself. The vacuoles continue to condense the proteins and the final
mature secretory granule is then moved to the membrane for secretion.
|
|
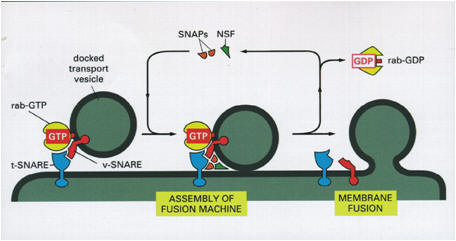
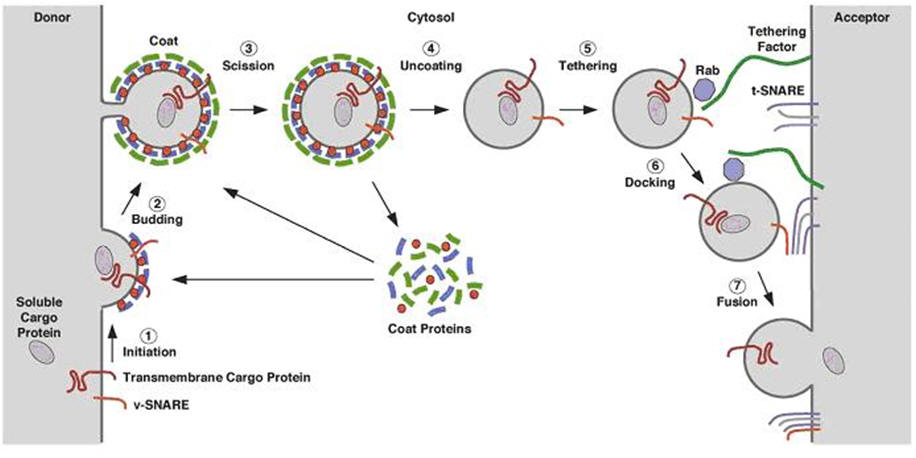
Transport of material in and out of the Golgi
complex thus involves budding and fusion of vesicles. The cartoon shows
that the membranes of each join and align themselves during the process
so that the inside face remains in the lumen and the outside face
remains towards the cytoplasm. |
|
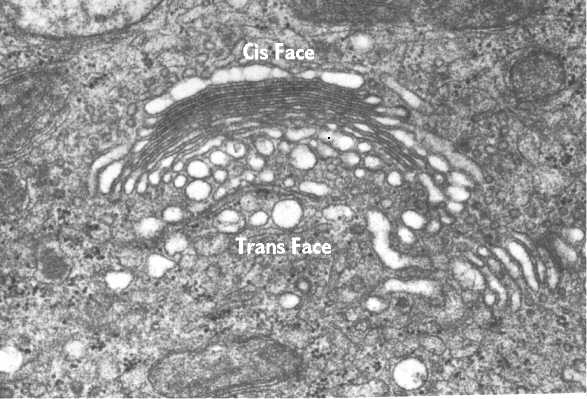
This
electron micrograph illustrates the Golgi complex. The Golgi is curved with its
trans face pointing away from the nucleus (top left) toward the cell periphery. The
numerous vesicles in the area are transporting the proteins to and from cisternae. |
 |
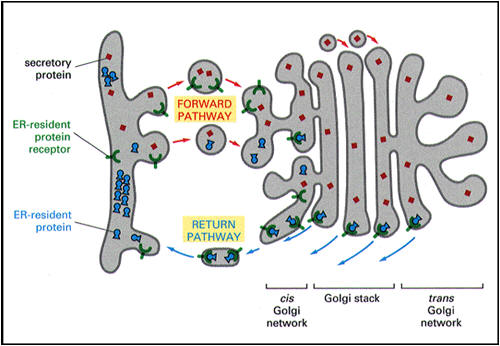
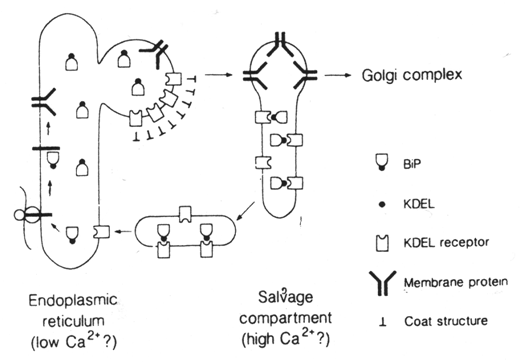
Can proteins be transported from the Golgi back to the ER?
|
Sometimes vital proteins needed in the ER are transported along with the
other proteins to the Golgi complex. The Golgi complex has a mechanism
for trapping them and sending them back to the ER. This cartoon shows
the process. The protein destined for secretion is red (travels via the
forward or anterograde pathway). The blue protein must remain in the ER.
The ER has inserted a receptor protein on the membrane it sends to the
Golgi complex in the transitional vesicles (the so-called KDEL receptor,
shown in green). These are retrograde vesicles and are therefore coated
with COPI. The KDEL receptor captures all of the protein that
carries the ER residency signal (the KDEL signal, dot in figure on the
right, e.g., in BiP). Vesicles then bud
from the Golgi complex and move back to the ER (retrograde pathway). The
receptor can circulate and continue to return the proteins needed by the
ER. |
|
 |
|
|
|
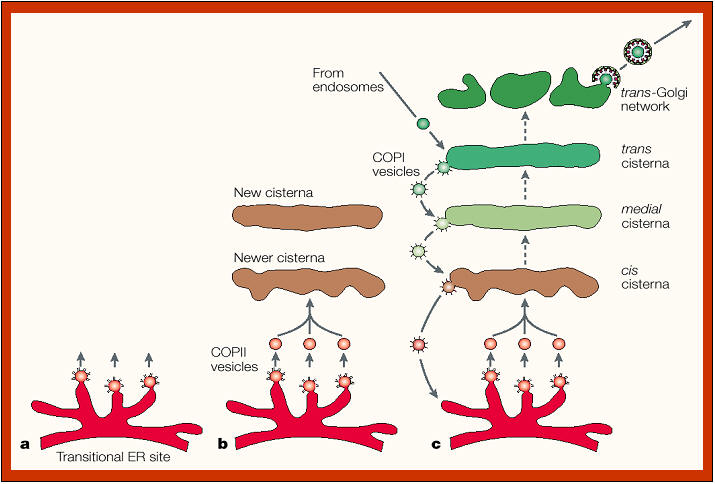
Golgi formation and cisternal
maturation
|
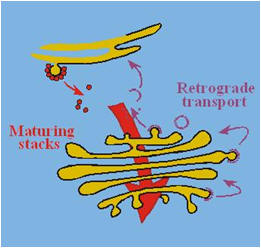
There is much interest in understanding how the different Golgi
cisternae are organized and differentiated. Two main models exist.
In the
“vesicular model” of protein transport (see figure above), all anterograde (forward) and
retrograde (backward) intra-Golgi steps involve transport only via
vesicles. Alternatively, a nonvesicular transport mechanism may be
present in which Golgi cisternae form at the cis-face of the stack,
probably by VTC fusion, and then progressively mature into trans-cisternae
(“cisternal maturation model”; see figures on the right and below). In this model, cisternae (or intermittent tubular continuities) carry secretory cargo
through the stack in the anterograde direction, while vesicles transport
Golgi enzymes in the retrograde direction, allowing cisternal maturation
to occur by progressive uptake of material from older stacks. At the TGN,
the cisternae ultimately disintegrate and evolve into a collection of
secretory vesicles, including immature secretory granules. |
 |
|
 |
 |
|
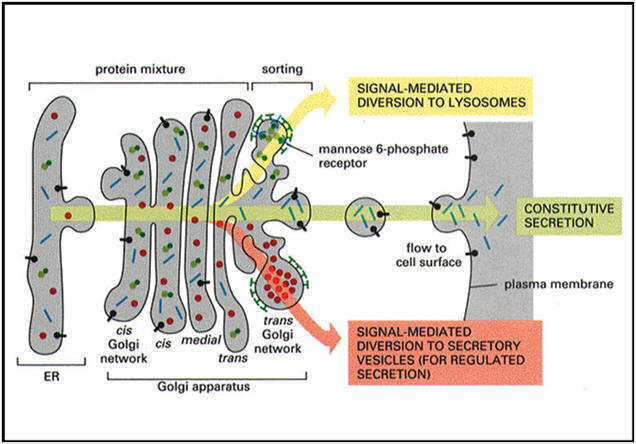
The
Golgi complex controls trafficking of different types of proteins. Some
are destined for secretion. Others are destined for the extracellular
matrix. Finally, other proteins, such as lysosomal enzymes, may need to
be sorted and sequestered from the remaining constituents because of
their potential destructive effects. The figures below show
the two types of secretory pathways. The regulated secretory pathway, as
its name implies, is a pathway for proteins that requires a stimulus or
trigger to elicit secretion. Some stimuli regulate synthesis of the
protein as well as its release. The constitutive pathway allows for
secretion of proteins that are needed outside the cell, like in the
extracellular matrix. It does not require stimuli, although growth
factors may enhance the process. Finally, the cartoons also show the
packaging of lysosomes. |
Golgi proteins
|
The Golgi apparatus
contains at least a thousand different types of integral and peripheral
membrane proteins, perhaps more than any other intracellular organelle.
The Golgi apparatus performs three major functions essential for growth,
homeostasis and division of eukaryotic cells. First, it operates as a
carbohydrate factory for the processing and modification of proteins and
lipids moving through the secretory pathway. Second, it serves as a
station for protein sorting and transport, receiving membrane from the
ER and delivering it to the plasma membrane or other intracellular
sites. Finally, it acts as a membrane scaffold onto which diverse
signaling, sorting and cytoskeleton proteins adhere.
These
distinct Golgi functions operate within a structure that is unique among
subcellular organelles in many ways, including its composition as a
stacked array of cisternae and connecting tubules/vesicles, its enormous
diversity of protein components (>1000 different types), and its
unrivaled capacity to dynamically transform in response to specific
stimuli or other cellular changes.
No class
of Golgi protein is stably associated with the Golgi. Integral membrane
proteins associated with the Golgi, including processing enzymes for
post-translational protein modification (i.e. mannosidase II, galactosyltransferase, etc), are continuously exiting
and re-entering the Golgi by membrane trafficking pathways leading to
and from the ER. Peripheral membrane proteins associated with the Golgi
exchange constantly between membrane and cytosolic pools. Newly
synthesized cargo proteins passing through the Golgi to other
destinations (which include both integral membrane and luminal proteins)
also spend relatively short periods of time in the Golgi. The residency
times for these different classes of Golgi proteins vary enormously:
Golgi processing enzymes stay for ~60 min, cargo proteins ~30 min, cargo
receptors ~10 min and peripheral proteins ~1 min.
|
|
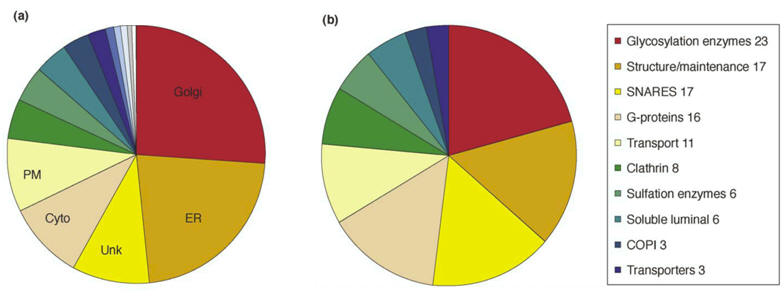
Classification of proteins identified in a rat liver Golgi proteome:
(a)
Proteins were grouped
by cellular location. Twenty-six percent of the identified proteins were
known Golgi proteins, whereas 23 percent were ER proteins. Many of the
cytosolic and cytoskeletal proteins functionally interact with the Golgi;
PM: plasma membrane, Unk: unknown.
(b)
Golgi-localized
proteins were grouped by function. |
|
Post-translational protein modifications in the Golgi complex
|
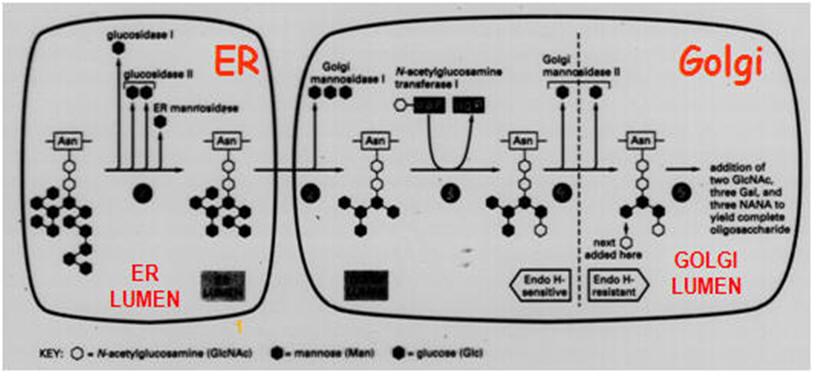
The Golgi
complex is compartmentalized. Phosphorylation occurs in the cis region.
In other regions, different types of carbohydrates are added as a
glycoprotein passes through the cisternae. The figure on the right illustrates the
different regions where sugars like mannose (man), galactose (gal), etc
are added. The final sorting is done in the trans-Golgi complex.
Proteins of all living organisms
are generally modified in many different ways. A functionally important
posttranslational modification is the phosphorylation of proteins. The
presence or absence of a phosphate group at specific hydroxyl amino
acids regulates the activity, stability, localization, and
oligomerization of proteins and in this way influences the flow through
metabolic pathways, the transduction of external and internal signals,
as well as the timing of developmental steps. The most complex and at
the same time energetically most costly protein modification is, however,
the glycosylation of proteins. |
 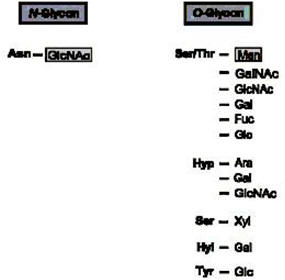
-
Types of sugar-peptide bonds in eukaryotes. Asn=asparagine;Ser
=serine;Thr =threonine; Hyl =hydroxylysine; Hyp =hydroxyproline; Tyr=tyrosine; GlcNAc
=N-acetylglucosamine; GalNAc =Nacetylgalactosamine; Glc
=glucose; Gal =galactose; Rha =rhamnose; Xyl=xylose; Ara =arabinose;
Man =mannose; Fuc=fucose.
|
|
Processing and maturation of N-glycan chains
Glycoproteins are proteins containing
covalently linked oligosaccharides that consist of different monomers
and are mostly branched. The carbohydrate moiety amounts to about 20% of
the molecular weight, but can be as much as 90% in some cases. In animal
cells, glycoproteins are distinguished from proteoglycans, extracellular
proteins with mostly long, unbranched polysaccharides, consisting of
serially repeating units. The so-called N-glycosylated proteins contain
oligosaccharides that are N-glycosidically linked to the g-amido group
of asparagines. This type of glycoprotein has been most intensively
studied with respect to their structure, biosynthesis, and function.
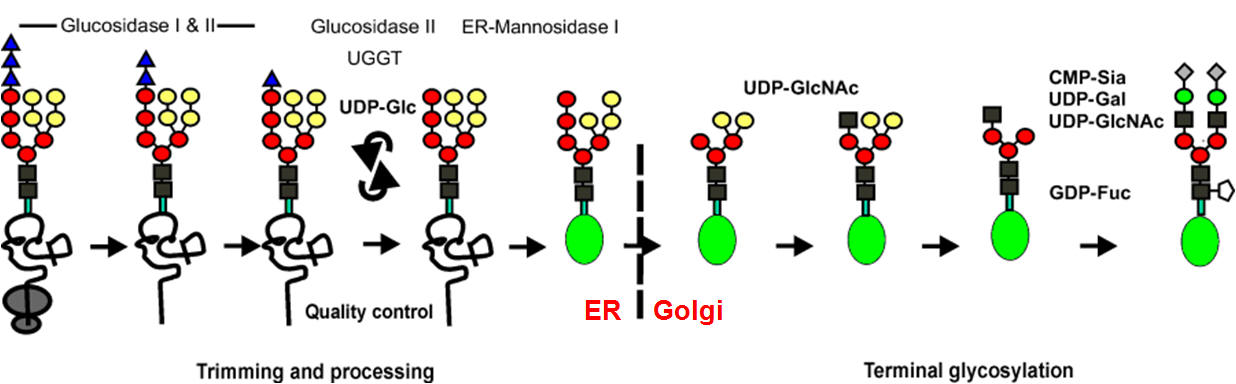
After the first
trimming steps in the ER, the exit of the correctly folded glycoprotein
(symbolized by a green ellipse, Figure below) occurs from the ER to the Golgi apparatus
where, in a strictly defined reaction sequence, a further demannosylation takes
place, followed by transfer of a GlcNAc residue, and finally removal of two
further mannose residues. In terminal glycosylation reactions, the mature glycan
structure is built up in a protein-dependent, tissue- and organism-specific
manner. The generated glycans are classified as high-mannose-type, complex-
type, and hybrid-type glycan structures. In the Figure below, only one possible terminal
pathway is depicted leading to a biantennary-complex-type glycan chain; the
number of antennae may vary up to six. In the case of soluble, lysosomal
glycoproteins, a mannose-6-phosphate determinant is generated that functions as
the signal for targeting the protein to the lysosome.
The many
different sugars, which are either N- or O-glycosidically linked to the
amino acid asparagine or to the hydroxy amino acids threonine, serine,
hydroxyproline, hydroxylysine, and tyrosine, reflect
the complexity of protein glycosylation. Protein
N-glycosylation and protein O-mannosylation are evolutionarily conserved
from yeast to man. In addition, there is a large
variety of more or less highly branched oligo- and polysaccharides of
varying composition that are linked to the proximal sugar of the
protein. The functional importance of protein glycosylation, however,
remained poorly understood for a long time, apart from the role of
saccharides as blood-group antigens. Only within the last few years
has it become increasingly evident that the lack of individual glycosyl
transferases contributing to the synthesis of sugar “trees” of specific
proteins can cause most severe congenital diseases in children,
including the CDG syndrome (congenital disorders of glycosylation) as
well as congenital muscular dystrophies with neuronal-cell-migration
defects. Although
the molecular details leading to these diseases are only vaguely
understood, it seems clear that sugar components of proteins play a
major role in embryonic and postembryonic development of humans as well
as of all higher eukaryotes.
|
|
 |
|
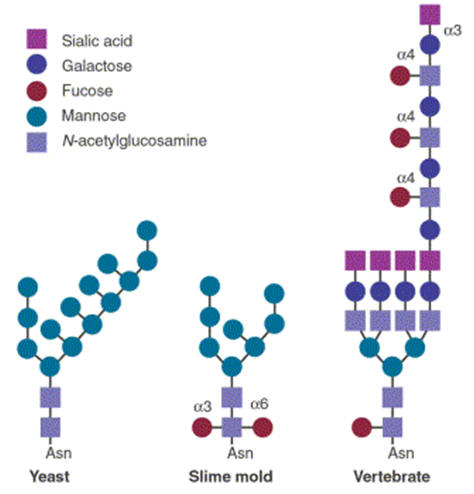
Glycoproteins processed by the Golgi apparatus, illustrating species
variation. |
See the movies "Protein
trafficking in the Golgi", "Protein
modifications in the Golgi", "Constitutive
secretion" and "Regulated
secretion" (use Quicktime).
Golgi maintenance and
biogenesis
The
Golgi apparatus contains thousands of different types of integral and
peripheral membrane proteins, perhaps more than any other intracellular
organelle. To understand these proteins' roles in Golgi function and in
broader cellular processes, it is useful to categorize them according to
their contribution to Golgi creation and maintenance. This is because
all of the Golgi's functions derive from its ability to maintain
steady-state pools of particular proteins and lipids, which in turn
relies on the Golgi's dynamic character - that is, its ongoing state of
transformation and outgrowth from the ER.
The Golgi
apparatus performs three major functions essential for growth,
homeostasis and division of eukaryotic cells. First, it operates as a
carbohydrate factory for the processing and modification of proteins and
lipids moving through the secretory pathway. Second, it serves as a
station for protein sorting and transport, receiving membrane from the
ER and delivering it to the plasma membrane or other intracellular
sites. Finally, it acts as a membrane scaffold onto which diverse
signaling, sorting and cytoskeleton proteins adhere.
These distinct
Golgi functions operate within a structure that is unique among
subcellular organelles in many ways, including its composition as a
stacked array of cisternae and connecting tubules/vesicles, its enormous
diversity of protein components (>1000 different types), and its
unrivaled capacity to dynamically transform in response to specific
stimuli or other cellular changes. Examples of the Golgi’s dynamic
behavior include its reversible disassembly during mitosis and under
experimentally induced conditions (e.g. osmotic stress or treatment with
BFA, Exo1 or ilimaquinone), and its rebuilding at peripheral ER export
sites in response to microtubule disruption or expression of mutated
proteins that function in ER-to-Golgi trafficking.
The Golgi’s
ability to transform itself fundamentally under different conditions is
probably due to the fact that proteins only associate with it
transiently as they move through other pathways in the cell. Conditions
that alter the entry or return of these proteins to the Golgi,
therefore, will disrupt Golgi structure and function. Also, many
proteins associated with the Golgi are part of large protein complexes.
Altering the association of one protein in the complex may affect the
stability and localization of others, with downstream consequences for
Golgi organization and structure.
Integral
membrane proteins associated with the Golgi are continuously exiting and
re-entering the Golgi by membrane trafficking pathways leading to and
from the ER. Peripheral membrane proteins associated with the Golgi, by
contrast, exchange constantly between membrane and cytosolic pools.
Newly synthesized cargo proteins passing through the Golgi to other
destinations (which include both integral membrane and luminal proteins)
also spend relatively short periods of time in the Golgi. The residency
times for these different classes of Golgi proteins vary enormously:
Golgi processing enzymes stay for ~60 min, cargo proteins ~30 min, cargo
receptors ~10 min and peripheral proteins ~1 min.
Post-translational
modifications of prohormones
In
addition to glycosylation and phosphorylation in the early stages of the
secretory pathway, a secretory protein can undergo sulfation at
carbohydrate side chains or tyrosine residues in the late pathway (by
carbohydrate and tyrosylprotein sulfotransferases,
respectively). Furthermore, the protein can undergo proteolytic
cleavage. The first step in proprotein proteolytic processing is usually
an endoproteolytic cleavage in the trans-Golgi network (TGN) or
immature secretory granule (ISG) on the carboxy-terminal
side of a recognition site, often a pair of basic amino acids and mostly
Lys-Arg or Arg-Arg. The proprotein processing enzymes are calcium- and
pH-dependent serine endoproteases related to the bacterial proteolytic
enzyme subtilisin and are called proprotein convertases (PCs). These
enzymes are structurally related (strongest in their active sites) and
form a gene family consisting of at least seven members, including furin
(also called PACE, Paired basic Amino acid residue Cleaving Enzyme), the
prohormone convertases PC1/PC3 and PC2, and (in yeast) KEX2. Furin and
KEX2 are active in constitutively secreting cells. The neuroendocrine-specific
enzymes PC1/PC3 and PC2 cleave prohormones and are selectively present
in cells equipped with the regulated secretory pathway. A differential
expression of PC1/PC3 and PC2 may result in a different secretory output
from endocrine and neuronal cells. Furthermore, a single prohormone may
give rise to multiple peptide hormones with a variety of bioactivities.
For example, processing of proopiomelanocortin (POMC) can result in
a-,
b- and
g-melanocyte-stimulating
hormones (MSHs), the stress hormone adrenocorticotropin (ACTH) and the
endorphins, peptides with endogenous opiate-like activity. POMC cleavage
by the PC1/PC3 enzyme generates ACTH, whereas PC2 produces the MSHs and
endorphins. PCs become active by autocatalytic cleavage of an
amino-terminal propeptide that may act as an intramolecular chaperone
for proenzyme folding. The 7B2 protein has been found to act as a
chaperone specific for PC2 by transiently interacting with the proenzyme
form, facilitating proPC2 transport and activation. The proSAAS protein,
with a structural organization similar to 7B2, appears to have PC1/PC3
as its major intracellular binding target. The proper acidic environment
in the subcompartments of the secretory pathway, essential for optimal
PC cleavage activity, is supplied by a H+-pumping
vacuolar-type ATPase (V-ATPase). Following cleavage by PCs,
exoproteolytic removal of the exposed carboxy-terminal basic residues
occurs by the enzyme carboxypeptidase E (or KEX1 in yeast). Finally,
the generated peptide may undergo one or two modifications that are
crucial for its biological activity, namely acetylation at the amino
terminus and amidation at the carboxy terminus if the peptide ends in
glycine (by the enzyme Peptidyl-glycine-α-Amidating
Monooxygenase, PAM). |
|
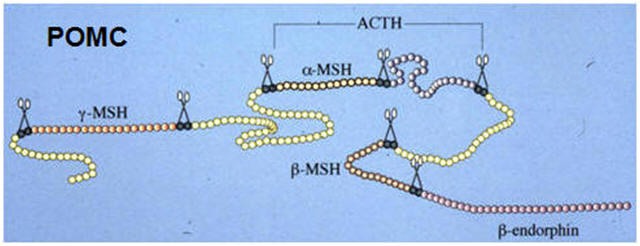 |
Proteolytic processing of the prohormones proinsulin and
proopiomelanocortin (POMC) |
|
Exocytosis
and endocytosis
Secretory proteins are released into the
extracellular space by exocytosis, a process involving the fusion of the
secretory granule membrane with the cell membrane. For the two types of
exocytosis, namely regulated (triggered) and constitutive (untriggered)
exocytosis, regulatory mechanisms of membrane fusion might be quite
different. Regulated exocytosis is coupled in most cases with
endocytosis to provide a membrane shuttle in order to prevent that the
plasma membrane would become too large which would occur when only
exocytotic events take place. A number of components of the exocytotic
and endocytotic machinery has been recently isolated and identified,
e.g. synaptophysin, synaptotagmin, synaptobrevin (VAMP), syntaxin and
annexins.
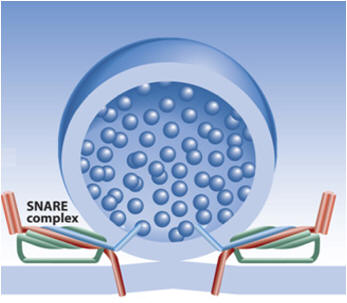
Exocytosis
Exocytosis is
thus the process whereby intracellular fluid-filled vesicles fuse with
the plasma membrane (PM), incorporating vesicle proteins and lipids into
the plasma membrane and releasing vesicle contents into the
extracellular milieu. This membrane fusion event thus mediates the
targeting of proteins and lipids to the PM and the secretion of
molecules from the cell. Exocytosis can occur constitutively or can be
tightly regulated, for example, neurotransmitter release from nerve
endings. The last two decades have witnessed the identification of a
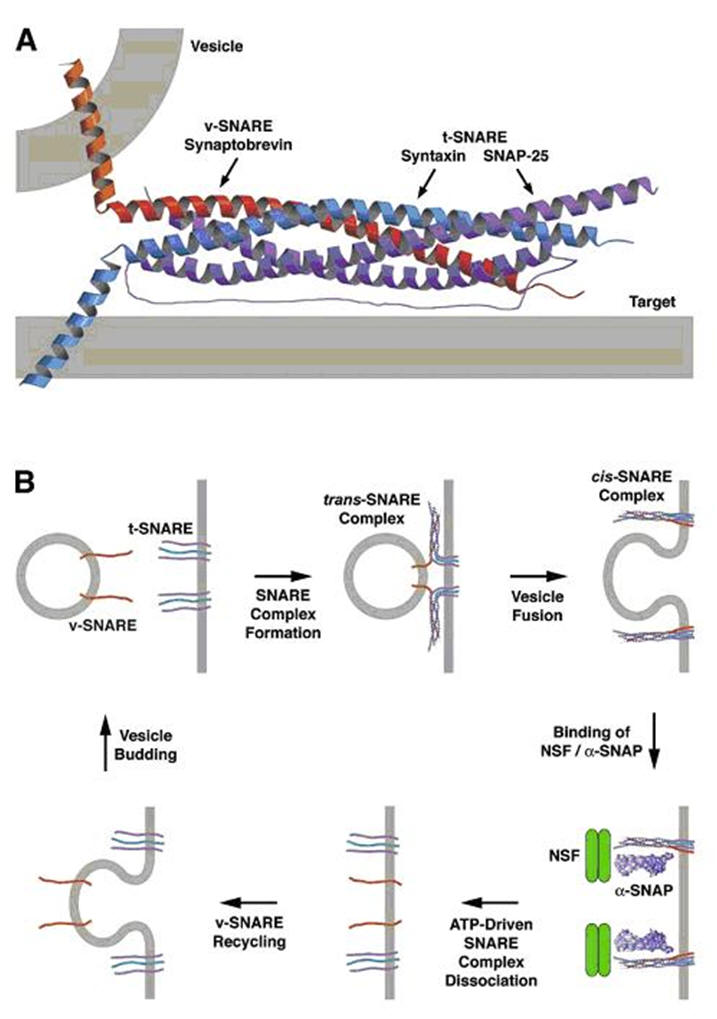
vast array of proteins and protein complexes essential for exocytosis.
As mentioned above, SNARE proteins are probable mediators of membrane
fusion, whereas other proteins function as essential SNARE regulators. A
central question that remains unanswered is how exocytic proteins and
protein complexes are spatially regulated.
Constitutive
exocytosis events include the fusion of vesicles derived from the
trans-Golgi network (TGN) with the PM, which is essential for the
insertion of newly synthesized proteins and lipids into the PM.
Polarized cells have developed specialized mechanisms for the targeting
of these TGN-derived vesicles to specific regions of the PM, for
example, apical vs. basolateral membrane in polarized epithelial cells.
In addition, proteins that are constitutively recycled through the
endosomal system, such as the transferrin receptor, are transported to
the cell surface via the fusion of endosomal vesicles with the PM. These
constitutive pathways operate in all cells. In addition, a number of
cell types undergo a more specialized form of exocytosis known as
regulated exocytosis. Exocytosis of regulated secretory vesicles only
occurs upon receipt of a specific stimulus, such as exocytosis of
synaptic vesicles in nerve cells. In the majority of cases, regulated
exocytosis is stimulated by a local and transient increase in calcium
levels.
Exocytosis can
involve the full fusion of a vesicle with the PM or, in more specialized
cases such as regulated exocytosis from neuronal and neuroendocrine
cells, can also occur by a "kiss-and-run" mechanism. Kiss-and-run
exocytosis involves the formation of a transient fusion pore that allows
release of a limited amount of the vesicle content before the pore
re-seals and the vesicle is released from the plasma membrane.
The study of
regulated exocytosis at the molecular level has been driven by a number
of key questions: How are vesicles recruited to the PM? What proteins
anchor vesicles to the PM? What proteins sense and respond to elevated
calcium levels? What proteins initiate and catalyze membrane fusion?
Finally, how are all these proteins regulated? These questions have led
to the identification of numerous proteins, each with their own
intricate contribution to exocytosis (see figure). The high efficiency of the
exocytosis machinery is exemplified in the ultra-fast response it can
exhibit to appropriate stimuli; for example, synaptic vesicles can fuse
with the presynaptic PM microseconds following calcium influx.
Lipid
rafts:
membrane platforms regulating intracellular pathways
Recent studies suggest that lipid
rafts, cholesterol and sphingolipid-rich microdomains, enriched in the
plasma membrane, play an essential role in regulated exocytosis
pathways. The association of SNAREs with lipid rafts acts to concentrate
these proteins at defined sites of the plasma membrane. Furthermore,
cholesterol depletion inhibits regulated exocytosis, suggesting that
lipid raft domains play a key role in the regulation of exocytosis. The
role of lipid rafts in regulated exocytosis can vary from a passive role
as spatial coordinator of exocytic proteins to a direct role in the
membrane fusion reaction.
The lipids that
compose cellular membranes are diverse, and as such have different
affinities towards proteins and other lipids. The lipid "raft"
hypothesis suggests that sphingolipids and cholesterol cluster into
discrete regions of the cell membrane. These sphingolipid- and
cholesterol-rich domains have been termed lipid "rafts" because they
exist in a less fluid and more ordered state than glycerophospholipid-rich
domains of the membrane. Studies on model membranes clearly demonstrate
clustering and segregation of sphingolipids, cholesterol and certain
types of glycerophospholipids, and there is now also evidence that
raft-type domains exist in living cells.
Lipid rafts are
resistant to solubilization by cold nonionic detergents; this resistance
has been used as the criterion for raft purification from numerous cell
types, and has allowed a detailed analysis of raft function in various
cellular pathways. The term "lipid raft" thus refers to membrane domains
that are resistant to detergent extraction. In addition to the
characterization of detergent-insoluble lipid rafts, fluorescent imaging
techniques such as fluorescence energy transfer and patching/co-patching
of membrane proteins have provided essential data on the domain
structure of the plasma membrane in fixed and living cells.
The ability of
lipid rafts to sequester specific proteins and to exclude others makes
them ideally suited to spatially organize cellular pathways. Rafts have
been implicated in the regulation of a range of signal transduction
pathways, where raft association of components of a signaling cascade
likely facilitates protein protein interactions and signal amplification.
Rafts have also been implicated in membrane traffic pathways, such as
the formation of regulated and constitutive secretory vesicles. protein interactions and signal amplification.
Rafts have also been implicated in membrane traffic pathways, such as
the formation of regulated and constitutive secretory vesicles.
Evidence that
lipid rafts play a key role in regulated exocytosis has emerged from a
number of recent studies examining the membrane domain distribution of
SNARE proteins. The conclusions form these studies:
SNARE proteins are clustered in the plasma membrane in a
cholesterol-dependent manner; a proportion of these cholesterol-rich
clusters correspond to lipid raft domains - the amount of SNAREs in
lipid raft domains depends upon the specific SNARE isoform and the cell
type; the integrity of lipid raft domains is important for exocytosis.
Rafts and
the regulation of membrane fusion and exocytosis
If SNAREs are
localized in different domains at the plasma membrane, what does this
mean for membrane fusion? Specifically, do the SNARE clusters in lipid
rafts or nonrafts mark the sites of exocytosis? Further analyses will
hopefully address whether regulated exocytosis occurs at specific
SNARE clusters and determine the molecular characteristics of these
fusion-competent domains.
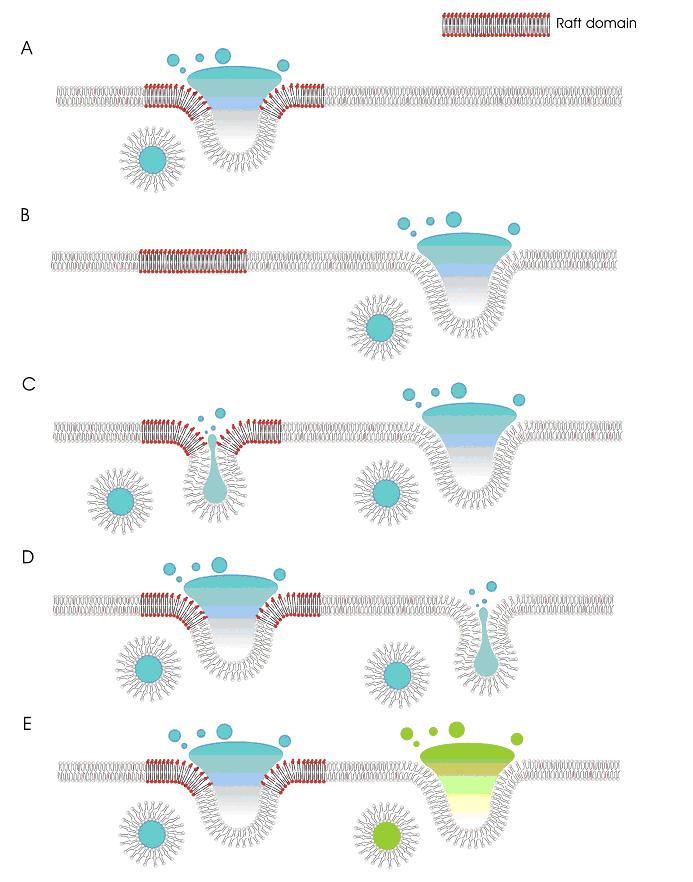
The
different protein and lipid composition of lipid rafts and nonraft
domains is likely to impact directly on membrane fusion and exocytosis.
There are a number of possibilities for how lipid rafts may function in
regulated exocytosis (Figure 5): The protein/lipid composition of rafts
is conducive to exocytosis, whereas membrane fusion in nonraft domains
is prevented (Figure 5A). The figure depicts fusion in the middle of the
lipid raft domain; alternatively, membrane fusion may occur at the edges
of raft domains, or rafts may surround the fusion site. Fusion occurs
exclusively in nonraft domains (Figure 5B). Lipid rafts and nonraft
domains support different types of exocytosis, for example full fusion
vs. kiss-and-run exocytosis. In the latter form of exocytosis, a
transient fusion pore is formed, allowing release of vesicle contents;
this fusion pore then quickly seals and the vesicle is released from the
plasma membrane. The lipid composition of rafts and nonraft domains may
favor one of these exocytic events (Figure 5C,D). Raft and nonraft
domains may also regulate the fusion of specific vesicle pools (Figure
5E) |
|
|
|