As we have seen, proteins are linear polymers synthesized by ribosomes from activated amino acids. The product of this biosynthetic process is a polypeptide chain, which has to adopt the unique three-dimensional structure required for its function in the cell. In 1972, Christian Anfinsen was awarded the Nobel Prize for Chemistry for showing that this folding process is autonomous in that it does not require any additional factors or input of energy. Based on in vitro experiments with purified proteins, it was suggested that the correct three-dimensional structure can form spontaneously in vivo once the newly synthesized protein leaves the ribosome. Furthermore, proteins were assumed to maintain their native conformation until they were degraded by specific enzymes. In the last decade, this view of cellular protein folding has changed considerably. It has become clear that a complicated and sophisticated machinery of proteins exists which assists protein folding and allows the functional state of proteins to be maintained under conditions in which they would normally unfold and aggregate. These proteins are collectively called molecular chaperones, because they prevent unwanted interactions between their immature clients. Molecular chaperones are a group of structurally diverse and mechanistically distinct proteins that either promote folding or prevent the aggregation of other proteins.
Click here for an animation on Protein folding. This animation illustrates how a protein folds from a straight chain of amino acids into a 3-dimensional object. Features are:
-
The spheres represent amino acids in a protein chain
-
Red spheres represent amino acids with hydrophobic side chains
-
Purple and blue spheres represent amino acids that can form hydrogen bonds with each other's side chains
-
Yellow and green spheres represent amino acids that can form ionic bonds with each other's side chains
-
-
Notice how the hydrophobic side chains start at different points along the chain but fold towards the center of the protein to avoid water
-
Similarly, the ionic and hydrogen bonds form between side chains to pull the protein into it's final shape.
-
Proteins most-likely fold by a process of 'trial and error"; they try various conformations until they find the most stable one.
-
Amino acid side chains can't move through each other.
Whereas many proteins can be refolded in vitro under optimized conditions in good yields, the situation in a living cell is less favorable. In particular, high protein concentration and temperature promote aggregation as an undesired side reaction, competing with productive folding. Unlike protein assembly, which describes the ordered association of several polypeptide chains into a defined functional oligomer, aggregation is the disordered, non-specific association of polypeptide chains that leads to the formation of heterogeneous protein particles devoid of any biological function. Considering the amount of energy the cell has already invested in the synthesis of a new polypeptide, it does not come as a surprise that strategies have evolved to promote the productive folding of a protein into its active conformation. During molecular evolution, polypeptide sequences were likely not only selected based on their biological properties, but also on whether they can fold productively.
In contrast to the situation in vitro, all proteins have to fold under the same set of conditions in a living cell. These conditions seem to be counterproductive for efficient folding, mainly because of the high temperature and the large number of non-native proteins present. Given the circumstances, it seems surprising that cells are usually devoid of aggregated proteins. There are two possible explanations for this observation. First, aggregation does occur in vivo, but its products are rapidly removed by cellular proteases. This would imply that cells waste a lot of energy to produce proteins that never become functional. Second, cells have found a strategy of minimizing the aggregation of newly synthesized proteins in the first place. As mentioned above, this has been achieved by the molecular chaperones, which influence the spontaneous folding reaction of proteins, thus preventing aggregation. It is important to note that the molecular chaperones do not provide specific steric information for the folding of the target protein, but rather inhibit unproductive interactions and thus allow the protein to fold more efficiently into its native structure.
Click here for an animation on molecular chaperones.
Molecular chaperones are found in all compartments of a cell where folding or, more generally, conformational rearrangements of proteins occur. Although protein synthesis is the major source of unfolded polypeptide chains, other processes can generate unfolded proteins as well. At non-physiological high temperatures or in the presence of certain chemicals, proteins can become structurally labile and may even unfold. Eventually, this would result in loss of function of the affected proteins and in the accumulation of protein aggregates. The cell responds to this threat by producing increasing amounts of specific protective proteins (heat-shock proteins, Hsps), a phenomenon referred to as heat-shock response or stress response. Many of these proteins were found to be molecular chaperones.
- Molecular chaperones:
- 1. Recognize nascent, unfolded, and partially folded proteins, & prevent misfolding and aggregation
- 2. Interact with and regulate the conformation of key regulatory proteins (transcription factors, receptors, caspases, kinases, phosphatases) and other activities that require the assembly and disassembly of heteromeric complexes
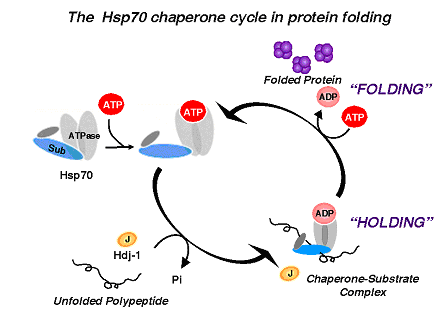
- 3. Often involve two phases: "holding" intermediate folded states and "folding" to the native state; folding requires ATP
- 4. Require co-chaperones to modulate holding and folding and confer substrate specificity
- 5. Have a key role in the triage decision between folding and degradation of damaged proteins
Functional properties of molecular chaperones
According to their molecular weight, molecular chaperones are divided into several classes or families. A cell may express multiple members of the same chaperone family. Proteins from the same class of molecular chaperones often show a significant amount of sequence homology and are structurally and functionally related, whereas there are hardly any homologies between chaperones from different families. Despite this diversity, however, most molecular chaperones share common functional features. The principal property of any molecular chaperone clearly is its ability to bind unfolded or partially folded polypeptides. During the early stages of folding or when misfolding occurs, the hydrophobic residues of a protein are partially solvent accessible and thus render it vulnerable to aggregation. Association of these hydrophobic protein species with molecular chaperones efficiently suppresses aggregation.
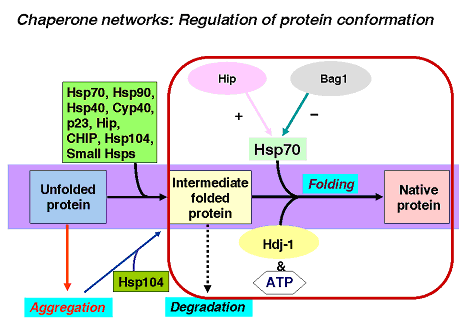
Chaperone networks
An important question that remains to be answered is how the different classes of molecular chaperones cooperate in the cell. Examples of such chaperone networks are the cooperation between the Hsp90 system and the Hsp70 system (Hsps with molecular weights of 90- and 70-kDa, respectively) during the activation of steroid hormone receptors. Another puzzling feature is the overlap in the function of the various chaperone classes. All chaperones are able to suppress the aggregation of folding proteins and are overproduced simultaneously under stress conditions. While cells may require some functional overlap for backup purposes, much remains to be learned about the specific roles of individual classes of chaperones.
Protein folding in the ER
The ER is a major protein folding compartment in a eukaryotic cell, second only to the cytosol. Many principles governing protein folding in the cytosol also apply to the ER, but the ER differs significantly from the cytosol topologically, in its chemical composition, and in its protein folding machinery. These differences can significantly affect protein folding in the ER. The ER is a membrane-surrounded compartment, and its luminal space is topologically equivalent to the extracellular space. Proteins destined for the ER are directed to this compartment through a predominantly hydrophobic signal sequence and have to, either co- or post-translationally, traverse the ER membrane.
As in the cytosol, the pH in the ER is near neutral. In mammalian cells, the ER is the major site for Ca2+ storage. A major function of the ER is thus to store calcium that is used for intracellular signaling in response to mitogenic and growth factor signal transduction pathways. ER luminal Ca2+ concentrations reach 5 mM, compared to 0.1mM in the cytosol. The majority of the ER-resident molecular chaperones and foldases are vigorous Ca2+-binding proteins. Perturbation of the ER Ca2+ pool affects their folding, activity and interactions with other chaperones.
N-linked glycosylation
A multitude of post-translational modifications occur in the ER: N-linked glycosylation, disulfide bond formation, lipidation, hydroxylation, oligomerization, etc. N-linked glycosylation is initiated by transfer of a core oligosaccharide to consensus Asn-X-Ser/Thr residues in the polypeptide chain (X can be any amino acid residue). Glycosylation serves several purposes in protein folding. First, due to the hydrophilic nature of carbohydrates, glycosylation increases the solubility of glycoproteins and defines the attachment area for the surface of the protein. Second, due to their large hydrated volume oligosaccharides shield the attachment area from surrounding proteins. Third, oligosaccharides interact with the peptide backbone and stabilize its conformation. Lastly, sequential trimming of sugar residues is monitored by a lectin machinery to report on the folding status of the protein. This calnexin/calreticulin cycle is one arm of the quality-control machinery in the ER that monitors protein conformations and dictates whether a molecule is exported to the Golgi or targeted for ER-associated degradation (ERAD; see also under “Protein degradation in the cell”).
Protein folding machinery
In order to travel along the secretory pathway and eventually reach their appropriate cellular destinations, newly synthesized secreted and membrane-bound proteins must fold and assemble correctly. Failure to do so thus results in their retention in the ER and eventual degradation. The proper conformational maturation of nascent secretory pathway proteins is both aided and monitored by a number of ER chaperones and folding enzymes in a complex process termed ER quality control. The protein folding machinery of the ER consists of three classes of proteins: foldases, molecular chaperones, and the lectins calnexin, calreticulin, and the ER degradation enhancing mannosidase-like protein EDEM. Foldases are enzymes that catalyze steps in protein folding to increase their rate. Prominent examples are cis–trans peptidyl–prolyl isomerases (PPIs/immunophilins) which catalyze the cis–trans isomerization of peptidyl–prolyl bonds and protein disulfide isomerases (PDIs). As mentioned above, molecular chaperones facilitate protein folding by shielding unfolded regions from surrounding proteins, they do not enhance the rate of protein folding, and they are classified into several groups: class HSP70 chaperones in the ER are BiP (glucose-regulated protein GRP78) and GRP170. BiP also participates in the translocation of nascent polypeptide chains into the ER. The HSP90 class chaperone GRP94/endoplasmin recognizes a subset of peptide substrates, in a manner coordinated with other chaperones, e.g. BiP, and facilitates the display of immunogenic peptides on MHC class I complexes. In addition, PDI has disulfide-dependent and -independent chaperone activity. Preferential interaction of unfolded proteins with ER-resident molecular chaperones constitutes the second arm of the quality-control machinery in the ER.
Click here for a movie on protein transport .
ER storage diseases
Diseases caused by malfunctioning of any aspect of the ER fall into one of the following classes (see Table 1).
I. Mutant cargo molecules: mutations affecting the fold of cargo molecules result in retention of the cargo in the ER.
II. A defective ER folding and transport machinery prevents wild-type proteins from reaching their destination: these can be very specific defects affecting just a single protein due to the fact that many specific client–chaperone pairs have evolved.
III. Defective unfolded protein response (UPR, see below) signaling: these diseases are caused by loss of one arm of the UPR, e.g. through mutation of a proximal or downstream gene involved in the UPR.
IV. Inhibition of adaptive responses regulated by the UPR: polyglutamine repeats cause proteasomal dysfunction, thus eliminating one arm of the UPR and subsequent activation of apoptosis.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Chaperones and disease
The importance of molecular chaperones for cell viability is illustrated by the fact that deletions of their genes are often lethal or cause severe cellular defects, such as reduced resistance to stress. If a specific chaperone activity is missing or reduced to subnormal levels, a disease may be the consequence. In other cases, the action of chaperones may cause a disease, as in the case of bovine spongiform encephalopathy or Creutzfeldt-Jakob syndrome, which are both characterized by the deposition of fibrillous aggregates of the prion protein (PrP) in the brain. For mammalian prion diseases, the involvement of chaperones is still speculative. In addition, chaperones are emerging as molecular targets for therapy in cancer, tissue transplantation, and septic shock syndrome, among others. Depending on their respective contribution, inhibition or stimulation of chaperone activity may be required for intervention.
- Molecular chaperones in biotechnology
- The production of recombinant proteins in bacteria is often impaired by the formation of inclusion bodies, which are large aggregates that consist mainly of inactive forms of the overexpressed protein. In some cases, this problem could be alleviated by the simultaneous overproduction of molecular chaperones. Molecular chaperones may thus become useful tools to optimize biotechnological processes and to establish fine-tuned cell factories for the production of recombinant proteins.
- Chaperones and evolution
- Small proteins usually do not require the assistance of molecular chaperones for in vitro folding, whereas larger proteins often do. Apparently, large proteins must confer certain evolutionary advantages on the cell, despite the additional expenses required for their production. Among others, two considerations may be important in this context: 1) The conformation space that is accessible for small proteins may be limited, that is, some three-dimensional structures can only be formed by longer polypeptide chains. Since the structure and the function of a protein are intimately related, some biological functions may be unique to large proteins; 2) Large proteins often comprise multiple domains, which have specific functions such as oligomerization, substrate binding, etc. This set-up allows the creation of proteins with novel properties by a simple rearrangement of existing building blocks. The intimate relationship between chaperones and evolution is exemplified the recently proposed function of Hsp90 during development. This chaperone may be essential to maintain the function of mutated proteins. Under stress conditions, this task can no longer be performed, and the mutations gain effect, thus resulting in new phenotypes and traits.
The unfolded protein response (UPR)
When protein folding in the ER is inhibited, signal transduction pathways, which increase the biosynthetic capacity and decrease the biosynthetic burden of the ER to maintain the homeostasis of this organelle, are activated. The ER has thus evolved functions to sense the stress in its lumen and transduce signals to the cytoplasm and the nucleus in an attempt to adapt for survival or otherwise induce apoptosis. These pathways/responses, including transcriptional activation and translational attenuation, are collectively termed the unfolded protein response (UPR). The UPR is characterized by a shutdown of global protein synthesis and activation of expression of genes coding for ER-resident proteins that are involved in the folding and processing reactions. Cell growth, differentiation, or stress may increase the protein folding load on the ER. In cell types with a high secretory load or an ER compartment that is uniquely susceptible to stress, the UPR becomes extremely important in cell survival and function. In general, the UPR is conserved across evolution and fundamentally important in cellular function and organismal development.
- The UPR and neurological disease
- Activation of the UPR is taken as an indicator that ER function is disturbed in a pathological process. Activation of the UPR has been found in various pathological states of the brain including ischemia and degenerative diseases. There is thus accumulating evidence that the UPR might play a role in the pathogenesis of a number of neurological diseases associated with the accumulation of abnormal (toxic) protein aggregates in and around affected neurons. The etiology of these diseases is related to impaired clearance, and the cellular response to the accumulating aggregated protein. Polyglutamine diseases and common neurodegenerative disorders, such as Alzheimer’s disease and Parkinson’s disease, represent a large class of conformational diseases associated with the accumulation of abnormal protein aggregates in and around affected neurons. The protein aggregates in these diseases are localized to the nucleus or the cytoplasm, yet trigger the UPR and promote cell death. Although different types of protein aggregates may occur in different cellular compartments, studies have demonstrated that accumulation of aggregates in the cytosol can inhibit the proteasome to prevent ERAD and thereby activate the UPR and subsequent apoptotic programs. Polyglutamine proteins are not only substrates for the ubiquitin–proteasome system, they may also serve as competitive inhibitors of this important pathway of intracellular protein degradation. The inhibition of the ubiquitin–proteasome system by polyglutamine proteins may effectively impair ERAD, thereby indirectly inducing the UPR. A better understanding of the function and activation of the UPR would provide means to manipulate this pathway in order to cure diseases associated with unfolded proteins.
- 1. Transient & prolonged appearance of intermediates
- 2. Appearance of misfolded protein structures and aggregates
- 3. Increased load on "protein quality control": shifting the balance on molecular chaperones and degradative machines
- 4. Induction of the Heat Shock Response to elevate the levels of molecular chaperones to restore homeostasis
- What happens if misfolded proteins and aggregates persist?
- 1. Proteotoxicity: formation of aggregates resulting in cellular dysfunctioning
- 2. "Gain of function" toxicity due to the association of key cellular proteins with misfolded proteins and aggregates
- 3. Misassembly of protein machines
- 4. Defects in cell growth and development: aberrant stress and signaling resulting in oncogenesis & cell death
- 5. Neurodegenerative disease