Proteins are continuously synthesised and degraded in all living organisms. The concentration of individual cellular proteins is determined by a balance between the rates of synthesis and degradation, which in turn are controlled by a series of regulated biochemical mechanisms. Differences in the rates of protein synthesis and breakdown result in cellular and tissue atrophy (loss of proteins from cells) and hypertrophy (increase in protein content of cells). The proper functioning of the cell thus requires careful control of the levels of important structural proteins, enzymes and regulatory proteins. The only way that cells can reduce the steady state level of a particular protein is by proteolytic degradation. Thus, complex and highly-regulated mechanisms have been evolved to accomplish this degradation. The degradation rates of proteins are important in determining their cellular concentrations. Proteins break down at rates ranging from 100% per hour to less than 10% per hour and their half-lives (time taken for loss of half the protein molecules) vary between 24 h and 72 h. Regulatory enzymes and regulatory proteins have much shorter half-lives of the order of 5-120 min. Protein breakdown can take place in the mitochondria, chloroplasts, the lumen of the endoplasmic reticulum (ER) and the endosomes, but occurs most commonly in one of two major sites of intracellular proteolysis: lysosomes and the cytosol. The individual degradation rates of proteins vary within a single organelle or cell compartment and also from compartment to compartment, due either to differing sensitivity to local proteases or differing rates of transfer to the cytosol or lysosomes. The range of protein degradation rates within a single organelle is limited, suggesting that the proteins may be treated as groups or families.
Short-lived regulatory proteins are degraded in the cytosol by local proteolytic mechanisms. All short-lived proteins are thought to contain recognition signals that mark them for early degradation. One commonly employed method is the selective labelling of targeted proteins by ubiquitin molecules (see also below). Ubiquitin, a protein of 76 amino acids, binds covalently to available lysine residues on target proteins, which are then recognised by proteases. A number of molecular recognition signals for intracellular protein degradation have been identified, and there are likely to be others as yet undiscovered. Additional degradative mechanisms exist for the identification and rapid degradation of proteins that contain translational or post-translational errors, or have been damaged in some way.
An additional role of intracellular proteolysis is in the stress-response. Cells that are subject to stress, such as starvation, heat-shock, chemical insult or mutation, respond by increasing the rates of proteolysis. One function of this enhanced proteolysis is to salvage amino acids from non-essential proteins. These amino acids can then be reutilized in the synthesis of essential proteins or metabolized directly to provide energy. Another function is in the repair of damage caused by the stress. For example, oxidative stress has been shown to damage a variety of proteins and cause them to be rapidly degraded. Some proteins are genetically unstable. There appear to be signal sequences which target proteins to degradation. These can be part of the protein structure, or added post-translationally.
The two major systems of protein degradation in the cell are the lysosomal pathway and the proteasomal pathway (including ER-Associated Protein Degradation, ERAD; see below).
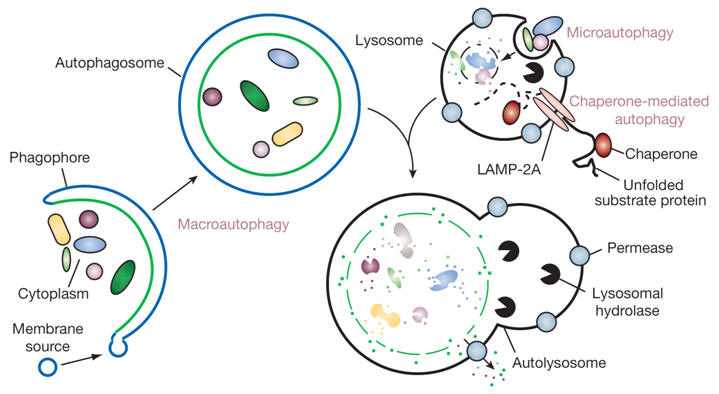
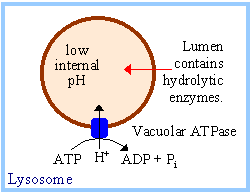
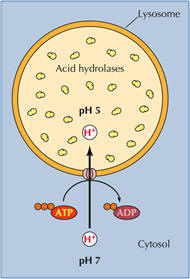
Lysosomal degradation. Most non-selective protein degradation takes place in the lysosomes, where changes in the supply of nutrients and growth factors can influence the rates of protein breakdown. Lysosomes contain a large variety of hydrolytic enzymes that degrade proteins and other substances taken in by endocytosis. Lysosomes have a low internal pH due to activity of vacuolar proton ATPase (see figure). All lysosomal hydrolases exhibit acidic pH optima.
Lysosomal proteases include many cathepsins (cysteine proteases), as well as some aspartate proteases and one zinc protease. Activation may be catalyzed by other lysosomal enzymes or be autocatalytic, promoted by the acidic pH within the lysosome.The rates of lysosomal degradation can vary greatly with cell type and conditions, ranging from less than 1% of total cell protein per hour to 5-10% per hour. The lysosomal degradation of some cytosolic proteins increases in cells deprived of nutrients. It is assumed that the proteins undergoing enhanced degradation are of limited importance for cell viability, and can be sacrificed to support the continuing synthesis of key proteins.
Proteins enter lysosomes by macroautophagy, that is the enclosure of a volume of the cytoplasm by an intracellular membrane. In autophagy, part of the cytoplasm may become surrounded by two concentric membranes. Fusion of the outer membrane of this autophagosome with a lysosomal vesicle results in degradation of enclosed cytoplasmic structures and macromolecules. Autophagy is not a mechanism for selective degradation of individual macromolecules.
Recognizing and destroying proteins that are unable to fold in the ER
A major function of the ER chaperones is to promote protein folding by preventing misfolding or aggregation. During conditions of ER stress, alterations in the ER environment can profoundly affect the folding of many proteins. Although BiP appears to be the sole chaperone that is monitored by the cell to sense ER stress, many of the chaperones are coordinately up-regulated. The main function of the increased levels of ER chaperones is to bind to unfolded proteins, prevent them from aggregating, and to aid and monitor their refolding if normal physiological conditions are restored to the ER. ER chaperones thus contribute to or control all of the major functions of the ER, including translocation of nascent polypeptide chains, folding and assembly of secretory pathway proteins, monitoring the success of this operation, identifying those proteins that fail this quality control and targeting them for degradation, and finally a role in storing calcium in the ER, which is important in cellular signal transduction pathways, and which also may play a role in initiating apoptosis when ER stress is sustained. Proteins that have ultimately failed ER quality control are degraded to prevent their accumulation in the ER. Accumulation might either titrate out the components of the chaperone systems or form large insoluble aggregates that would be toxic to the cell. The turnover mechanism is termed ERAD, which is conserved from lower eukaryotes like yeast to mammals. ERAD is a process by which misfolded ER proteins are detected and prevented from progressing along the secretory pathway, and directed to the translocon for retrotranslocation (or dislocation) into the cytosol, where they undergo ubiquitin- and proteasome-dependent degradation (cytoplasmic ubiquitin-proteasome system, UPS). Ubiquitin (Ub) is a highly conserved small protein that is universally expressed in eukaryotic cells. Ubiquitination of substrates is a multi-step process. Thus, a link exists between the turnover of ER membrane proteins and UPS. Retrotranslocation may utilize the same core protein complex that forms the protein conducting channel in the translocon through which proteins are delivered to the ER lumen. ERAD thus eliminates misfolded or unassembled proteins from the ER. ERAD targets are selected by a quality control system within the ER lumen (calnexin and BiP play sequential roles in identifying and targeting ERAD substrates for degradation) and are ultimately destroyed by the UPS.
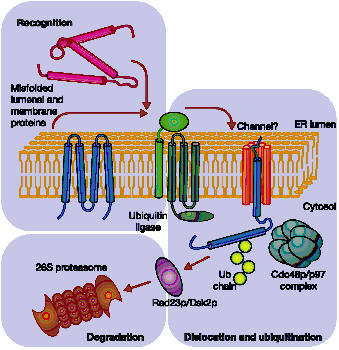
Proteasomal degradation of ERAD targets. Aberrant proteins are recognized within the ER lumen by different quality control mechanisms, which escort terminally misfolded polypeptides to a putative channel that facilitates their export from the ER. Cytoplasmically exposed lysine residues are ubiquitinated by ubiquitin ligases. Dislocation is completed with the help of a protein complex (Cdc48p/p97) and membrane-extracted substrates are conveyed to the proteasome by accessory factors (such as Rad23p and Dsk2p).
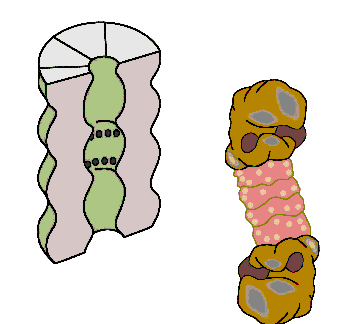
The cell's waste disposer, the proteasome. The black spots indicate active, protein-degrading surfaces.

Ubiquitin-mediated protein degradation
- The E1 enzyme activates the ubiquitin molecule. This reaction requires energy in the form of ATP.
- The ubiquitin molecule is transferred to a different enzyme, E2.
- The E3 enzyme can recognise the protein target which is to be destroyed. The E2-ubiquitin complex binds so near to the protein target that the actual ubiquitin label can be transferred from E2 to the target.
- The E3 enzyme now releases the ubiquitin-labelled protein.
- This last step is repeated until the protein has a short chain of ubiquitin molecules attached to itself.