By the early 1900s, geneticists understood that Mendel's laws of inheritance underlie the transmission of genes in diploid organisms. They noted that some traits are inherited according to Mendel's ratios, as a result of alterations in single genes, and they developed methods to map the genes responsible. They also recognized that most naturally occurring trait variation, while showing strong correlation among relatives, involves the action of multiple genes and nongenetic factors.
Although it was clear that these insights applied to humans as much as to fruit flies, it took most of the century to turn these concepts into practical tools for discovering genes contributing to human diseases. Starting in the 1980s, the use of naturally occurring DNA variation as markers to trace inheritance in families led to the discovery of thousands of genes for rare Mendelian diseases. Despite great hopes, the approach proved unsuccessful for common forms of human diseases—such as diabetes, heart disease, and cancer—that show complex inheritance in the general population.
Over the past year, a new approach to genetic mapping has yielded the first general progress toward mapping loci that influence susceptibility to common human diseases. Still, most of the genes and mutations underlying these findings remain to be defined, let alone understood, and it remains unclear how much of the heritability of common disease they explain.
Genetic mapping by linkage and association
Genetic mapping is the localization of genes underlying phenotypes
on the basis of correlation with DNA variation, without the
need for prior hypotheses about biological function. The
simplest form, called linkage analysis, was conceived for
fruit flies in 1913. Linkage analysis involves crosses between
parents that vary at a Mendelian trait and at many polymorphic
variants ("markers"); because of meiotic recombination, any
marker showing correlated segregation ("linkage") with the
trait must lie nearby in the genome.
In the 1970s, the ability to clone and sequence DNA made it possible to tie genetic linkage maps in model organisms to the underlying DNA sequence, and thereby to molecularly clone the genes responsible for any Mendelian trait solely on the basis of their genomic position. Such studies typically involved three steps: (i) identifying the locus responsible through a genome-wide search; (ii) sequencing the region in cases and controls to define causal mutation(s); and (iii) studying the molecular and cellular functions of the genes discovered. So-called "positional cloning" became a mainstay of experimental genetics, identifying pathways that are crucial in development and physiology.
Linkage analysis in humans
For most of the 20th century, genome-wide linkage mapping was impractical in humans: Family sizes are small, crosses are not by design, and there were too few classical genetic markers to systematically trace inheritance. Progress in identifying the genes contributing to human traits was initially limited to studies of biological candidates such as blood-type antigens and hemoglobin β protein in sickle-cell anemia.
In 1980,
Botstein and colleagues, building on their use of DNA
polymorphisms to study linkage in yeast and the finding
of DNA polymorphism at the globin locus in humans, proposed
the use of naturally occurring DNA sequence polymorphisms as
generic markers to create a human genetic map and
systematically trace the transmission of chromosomal
regions in families. The feasibility of genetic mapping
in humans was soon demonstrated with the localization of
Huntington disease in 1983. A rudimentary genetic linkage
map with ![]() 400
DNA markers was generated by 1987 and was fleshed out to
400
DNA markers was generated by 1987 and was fleshed out to ![]() 5000
markers by 1996. Physical maps providing access to linked
chromosomal regions were developed by 1995. With these
tools, positional cloning became possible in humans, and
the number of disorders tied to a specific gene grew from
5000
markers by 1996. Physical maps providing access to linked
chromosomal regions were developed by 1995. With these
tools, positional cloning became possible in humans, and
the number of disorders tied to a specific gene grew from ![]() 100
in the late 1980s to >2200 today.
100
in the late 1980s to >2200 today.
Several lessons emerged from studies of Mendelian disease genes: (i) The "candidate gene" approach was inadequate; most disease genes were completely unsuspected on the basis of previous knowledge. (ii) Disease-causing mutations often cause major changes in encoded proteins. (iii) Loci typically harbor many disease-causing alleles, mostly rare in the population. (iv) Mendelian diseases often revealed great complexity, such as locus heterogeneity, incomplete penetrance, and variable expressivity.
Geneticists were eager to apply genetic mapping to common diseases, which also show familial clustering. Mendelian subtypes of common diseases [such as breast cancer, hypertension, and diabetes] were elucidated, but mutations in these genes explained few cases in the population. In common forms of common disease, risk to relatives is lower than in Mendelian cases, and linkage studies with excellent power to detect a single causal gene yielded equivocal results.
These features were consistent with, but did not prove, a polygenic model. The idea that commonly varying traits might be polygenic in nature was offered in 1910. By 1920, linkage mapping was used to identify multiple unlinked factors influencing truncate wings in Drosophila, and a mathematical framework was developed for relating Mendelian factors and quantitative traits. In the late 1980s, linkage mapping of complex traits was made feasible for experimental organisms through the use of genetic mapping in large crosses. But there was little success in humans.
Genetic association in populations
A possible path forward emerged from population genetics and genomics. Instead of mapping disease genes by tracing transmission in families, one might localize them through association studies—that is, comparisons of frequencies of genetic variants among affected and unaffected individuals. Genetic association studies were not a new idea. In the 1950s, such studies revealed correlations between blood-group antigens and peptic ulcer disease; in the 1960s and 1970s, common variation at the human leukocyte antigen (HLA) locus was associated with autoimmune and infectious diseases; and in the 1980s, apolipoprotein E was implicated in the etiology of Alzheimer's disease. Still, only about a dozen extensively reproduced associations of common variants (outside the HLA locus) were identified in the 20th century.
A central problem was that association studies of candidate genes were a shot in the dark: they were limited to specific variants in biological candidate genes, each with a tiny a priori probability of being disease-causing. Moreover, association studies were susceptible to false positives due to population structure, because there was no way to assess differences in the genetic background of cases and controls. Although many claims of associations were published, the statistical support tended to be weak and few were subsequently replicated.
In the
mid-1990s, a systematic genome-wide approach to association
studies was proposed: to develop a catalog of
common human genetic variants and test the variants for association
to disease risk. The focus on common variants as a mapping
tool was a matter of practicality, grounded in population
genetics. The human population has recently grown
exponentially from a small size. Humans have
limited genetic variation: The heterozygosity rate for
single-nucleotide polymorphisms (SNPs) is ![]() 1
in 1000 bases. Moreover, perhaps 90% of heterozygous
sites in each individual are common variants, typically
shared among continental populations.
1
in 1000 bases. Moreover, perhaps 90% of heterozygous
sites in each individual are common variants, typically
shared among continental populations.
If most genetic variation in an individual is common, then why are mutations responsible for Mendelian diseases typically rare? One answer is natural selection: mutations that cause strongly deleterious phenotypes—as most Mendelian diseases appear to be—are lost to purifying selection. But if deleterious mutations are typically rare, how could common variants play a role in disease? Common diseases often have late onset, with modest or no obvious impact on reproductive fitness. Mildly deleterious alleles can rise to moderate frequency, particularly in populations that have undergone recent expansion. Moreover, some alleles that were advantageous or neutral during human evolution might now confer susceptibility to disease because of changes in living conditions accompanying civilization. Finally, disease-causing alleles could be maintained at high frequency if they were under balancing selection, with disease burden offset by a beneficial phenotype (as in sickle-cell disease and malaria resistance).
These lines of reasoning led to the so-called "common disease–common variant" (CD-CV) hypothesis: the proposal that common polymorphisms (classically defined as having a minor allele frequency of >1%) might contribute to susceptibility to common diseases. If so, genome-wide association studies (GWAS; see also under “GWAS”) of common variants might be used to map loci contributing to common diseases. The concept was not that all causal mutations at these genes should be common (to the contrary, a full spectrum of alleles is expected), only that some common variants exist and could be used to pinpoint loci for detailed study. It took a decade to develop the tools and methods required to test the CD-CV hypothesis: (i) catalogs of millions of common variants in the human population, (ii) techniques to genotype these variants in studies with thousands of patients, and (iii) an analytical framework to distinguish true associations from noise and artifacts.
Cataloging SNPs and linkage disequilibrium
Pilot
projects in the late 1990s showed that it was possible to
identify thousands of SNPs and to perform highly
multiplexed genotyping by means of DNA microarrays. A
public-private partnership, the SNP Consortium, built an
initial map of 1.4 million SNPs; this has grown to more
than 10 million SNPs and is estimated to contain 80% of
all SNPs with frequencies of >10%. As the SNP catalog grew, a
critical question loomed: would GWASs require directly
testing each of the ![]() 10
million common variants for association to disease? That
is, if only 5% of variants were tested, would 95% of
associations be missed? Or could a subset serve as
reliable proxies for their neighbors? Experience from
Mendelian diseases suggested that substantial efficiencies
might be possible. Each disease-causing mutation arises on a
particular copy of the human genome and bears a specific set
of common alleles in cis at nearby loci, termed a haplotype.
Because the recombination rate is low [
10
million common variants for association to disease? That
is, if only 5% of variants were tested, would 95% of
associations be missed? Or could a subset serve as
reliable proxies for their neighbors? Experience from
Mendelian diseases suggested that substantial efficiencies
might be possible. Each disease-causing mutation arises on a
particular copy of the human genome and bears a specific set
of common alleles in cis at nearby loci, termed a haplotype.
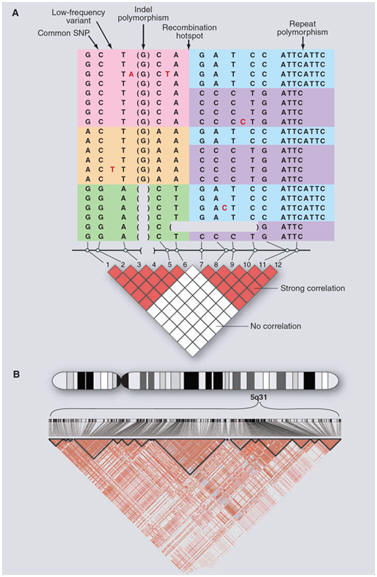
Because the recombination rate is low [![]() 1
crossover per 100 megabases (Mb) per generation], disease
alleles in the population typically show association with
nearby marker alleles for many generations, a phenomenon
termed linkage disequilibrium (LD) (Figure 1).
1
crossover per 100 megabases (Mb) per generation], disease
alleles in the population typically show association with
nearby marker alleles for many generations, a phenomenon
termed linkage disequilibrium (LD) (Figure 1).