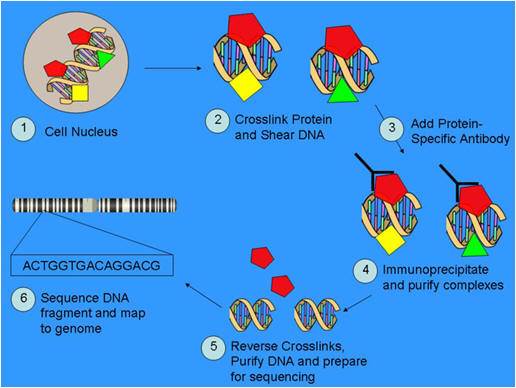
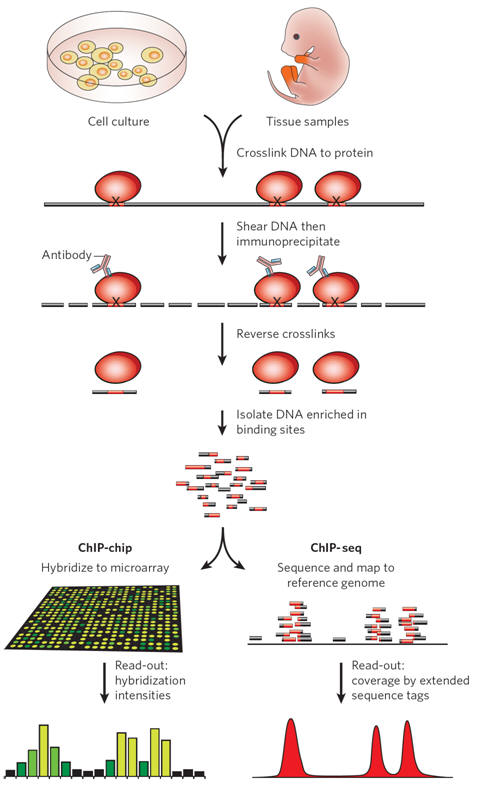
The ChIP technique involves formaldehyde crosslinking of DNA to proteins that bind to it directly or as part of larger complexes, combined with subsequent immunoprecipitation. This targeting of specific DNA-associated proteins was widely used in the pre-genomic era to study protein–DNA interactions directly in cultured cells or in tissue samples. The figure on the right and the top portion of Figure 1 shows a schematic overview of the individual steps involved. They include the molecular fixation of non-covalent protein–DNA interactions, shearing of the crosslinked chromatin, immunoprecipitation with an antibody binding the protein of interest and reversal of crosslinks. In many cases, antibodies that bind to covalently modified proteins are used, for example those that recognize methyl groups at defined amino-acid residues of histones. In the conventional ChIP approach, enrichment of the associated DNA fragments relative to non-immunoprecipitated ('input') DNA is quantified for individual proposed binding locations (not shown). This need for quantification at every site of interest initially thwarted the application of ChIP on a genomic scale.
The introduction of DNA microarrays allowed the hybridization-based interrogation of large numbers of potential binding sites in parallel (ChIP-on-chip, or ChIP-chip), thus making it possible to screen entire compact model-organism genomes or large vertebrate genome intervals in a single experiment (see Figure 1, bottom left). Recently, chromatin immunoprecipitation coupled to massively parallel sequencing (ChIP-seq) has become increasingly used as an alternative to ChIP-chip. The ChIP-seq method is very similar to the experimental set-up of ChIP-chip, except that, in the final step, next-generation sequencing techniques are used to determine the sequence of immunoprecipitated DNA fragments, which are then computationally mapped to the reference genome (see Figure 1, bottom right). Improved sequencing technologies offer the possibility to obtain millions of mappable reads in a single ChIP-seq experiment at moderate cost. The results from ChIP-seq are based on statistical analysis of read counts, which overcomes many of the challenges associated with the quantification and normalization of hybridization signals, and an increasing number of advanced computational ChIP-seq analysis tools are becoming available. ChIP-seq analysis covers by default the entire mappable portion of the reference genome without the need to restrict the analysis to its subregions.