A biological network is a set of molecules,
e.g. proteins or genes, that are linked
together via defined functional
relationships. The inter-connections between
molecules contain a wealth of information
that has yet to be fully exploited in
network-based analysis. Deciphering the
patterns of wiring in a system allows us to
penetrate the apparent complexity, and
understand how these wirings could result in
coordinated function. Early discoveries
suggest that biological networks share
common properties with many other natural
and man-made systems. For example, highly
connected proteins (hubs) are more likely to
be essential for cellular survival.
Bioinformatics is the use of computer-based tools to assist in the analysis of biological data. The development of DNA microarray and sequencing techniques, and advances within these fields has allowed a vast amount of data to be obtained in a short space of time (most notably from the Human Genome Project). This has coincided with technological developments within the computer industry. Computer-based tools (in silico analysis) can be used in a variety of ways to analyse biological data, as illustrated below.
Similarity searches
Similarity searches are the most common bioinformatics-based tools and are located on many bioinformatics based websites e.g. the Basic Local Alignment Search Tool (BLAST). These tools allow the comparison of sequences that have no assigned function against ones that do. The websites typically contain a user interface allowing the user to input a sequence. The search algorithm then compares this sequence (unknown function) with all the sequences held in a database (known function) and assigns a similarity value. This allows a potential function to be assigned to the sequence in a considerably shorter time when compared to laboratory methods. The results of this type of search could potentially dictate future research avenues within the laboratory.
A comparison between two sequences must take into account certain points. If a query sequence will match with a higher degree of homology when a 'gap' is inserted, then the algorithm will perform the insertion. Scoring penalties are employed to reduce the number of gaps at different points along with length of gaps employed. All possible comparisons are taken into account and a final statistical value is given. This determines how similar the two sequences are. The hypothesis is that if the sequences are similar, then there is a good chance the function will also be. Similarity searches can be carried out for amino acid sequences as well as DNA sequences. In practise, both DNA and protein databases are searched and due to the degeneracy of the genetic code, different results may be obtained. As more organisms are sequenced and the databases become more reliable, so the results will become more accurate. The main role of genome databases is to distribute information for published or finished work. The databases are open to the public. Different types of databases exist that can be used for the analysis and functional characterisation of sequences. When considering DNA databases the main contributors are a combination of three sites, Genbank (US), EMBL (European), and DDBJ (Japan). Organism-specific databases are also common.
Performing secondary database searches, allowing conserved motifs to be identified, is a supplement to primary DNA/Protein database similarity searching. This type of search allows further characterisation of a sequence. Once similarity and secondary searches have been carried out, multiple alignment tools are used. Comparing multiple alignments allows the possibility of conserved sequences to be identified. These are also termed 'motifs'. Identification of such conserved sequences represents possible structural and functional similarities and also possible evolutionary links between sequences. This procedure may at first seem to be pointless if initial primary and secondary sequence searches have produced insignificant results. However, if a number of sequences are aligned simultaneously, important gene families (conserved domains) can be identified.
Functional assignment
Genome sequencing has led to a vast amount of information being deposited onto databases. One of the most important ways in which these sequences can be used is to try and assign function to them. Functional assignment is a large topic in bioinformatics and has many software tools that can be applied to this subsection of bioinformatics. There are two main techniques that can be employed to aid the process of functional assignment:
1. Use sequence-based methods, typically involving similarity search methods as described above.
2. Use structure-based methods, involving the use of bioinformatics software that predicts 3D protein structure from sequence. These tools allow users to input sequences into the program and then return a predicted structure that can be viewed in a viewing software and the hypothetical structures can then be compared to known structures held in a structure database with the aim of identifying structural homologues. Thus, with more and more sequences being made available, this approach is getting more and more attractive.
Phylogenetic analysis
Identification of similar sequences can also lead to phylogenetic analysis. Programs calculate the best possible phylogenetic tree diagram. Sequences containing homology could have diverged through evolution. Computational phylogenetic analysis can be a very useful supplementary tool for the characterisation of a sequence if used correctly.
Software-based tools for specific applications
An alternative bioinformatics-based tool is to use computer software designed for specific applications, e.g. to analyse microarray results in order to identify up- and downregulated genes within microarray experiments (quantification of spots on the microarray). Once the arrays have been scanned and quantification has been carried out, the next step is to normalize the data. Experiment normalizations are used to standardize microarray data to enable differentiation between real (biological) variations in gene expression levels and variations due to the measurement process. Normalizing also scales data such that one can compare relative gene expression levels.
Network and pathway analysis
The biological system is a complex physicochemical system consisting of numerous dynamic networks of biochemical reactions and signaling interactions between cellular components. This complexity makes it virtually unanalyzable by traditional methods. Hence, biological networks have been developed as a platform for integrating information from high- to low-throughput experiments for analysis of biological systems. The network analysis approach is vital for successful quantitative modeling of biological systems. The advantage of network-based analysis methods is that, rather than using pathways as a whole, they identify subnetworks that are significantly differentially expressed.
A biological network is a simplified model
that describes the inter-relationships
between a set of functional entities such as
genes, proteins or metabolites: metabolic
pathways (MNs), regulatory pathways (RNs),
protein–protein interactions (PPINs),
genetic interactions (GINs), protein
complexes and proteins annotated to the same
Gene Ontology (GO) terms. MNs link two
proteins in a directed relationship if the
product of one is the substrate of the other.
RNs refer to transcriptional relationships
or other indirect relationships where one
protein controls the expression or
repression of the other. MNs and RNs are
thus natural biological pathways. In PPINs,
a relationship between two proteins exists
if they are experimentally verified to
interact physically. In GINs, a gene
interacts with another if a combined
mutation between them results in a more
severe phenotype as opposed to a single
mutation in either of them. A GIN may imply
a physical interaction (as part of a
complex) or a complete ablation of functions
across two compensatory pathways. GINs are
only beginning to be better understood but
remain difficult to study empirically.
Unlike MNs and RNs, PPINs and GINs are
purely pairwise interaction information and
cannot yet be put into the context of a
natural biological pathway.
The GO was established by the Gene Ontology
Consortium as an important reference
terminology for annotating the function and
cellular localization of proteins. GO terms
are organized into three separate
hierarchical ontologies — viz., cellular
component terms (CC), molecular function
terms (MF) and biological process terms
(BP). Associated with the GO is a large and
well-organized database of proteins
annotated to GO terms. In particular, when a
group of proteins are annotated to a CC, BP
or MF term, it means that this group of
proteins is localized to that cellular
compartment (corresponding to the CC term),
participate in that biological process (corresponding
to the BP term), or participate in that
molecular function (corresponding to the MF
term), respectively. Protein complexes and
proteins annotated to the same GO terms are
not actually networks. Nevertheless,
proteins that are in the same complex or
annotated to the same GO terms are
functionally linked and can be considered to
form functional linkage networks.
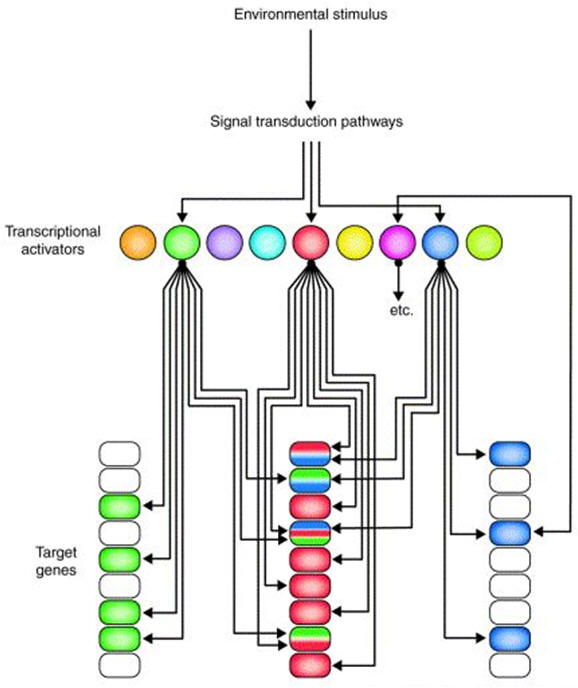 Gene
expression regulatory network
Gene
expression regulatory network
See also: Molecular networks.