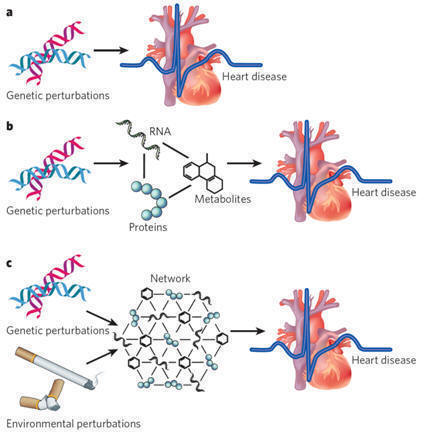
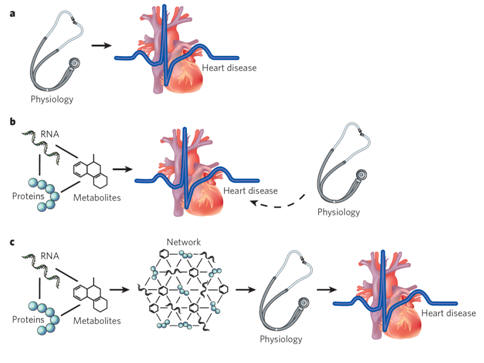
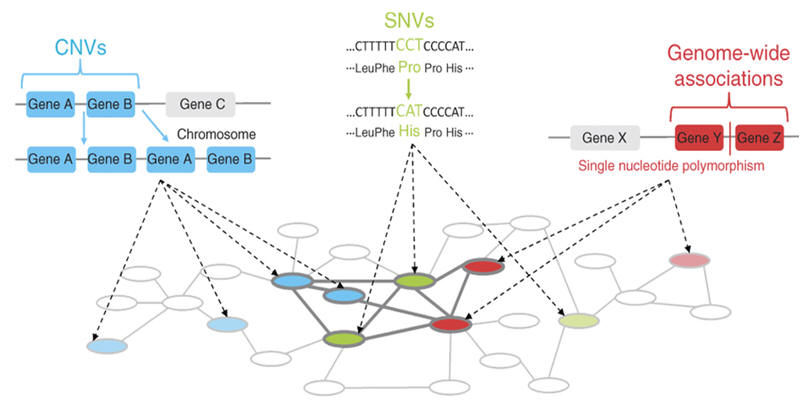
|
Perspectives
The
disease-associated molecular networks that can be constructed today are
necessarily based on grossly incomplete sets of data. Even given the
ability to assay DNA and RNA variation in whole populations in a
comprehensive manner, the information is not complete, because we are
far from completely characterizing rare variation, DNA variation other
than SNP and copy number, variation in non-coding RNA levels and
variation in the different isoforms of genes in any sample, much less in
entire populations. Beyond DNA and RNA, it is not possible with existing
technologies to measure all protein-associated traits or all the
interactions between proteins and DNA/RNA, metabolite levels and other
molecular entities important to the functioning of living systems.
Furthermore, the types of high-dimensional data we are able to generate
routinely today in populations represent only a snapshot at a single
time point, which may allow the identification of the functional units
of the system under study and how these units relate to one another but
does not allow a complete understanding of how the functional units are
put together or the mechanistic underpinnings of the complex set of
functions carried out by individual cells, by entire organs and by whole
systems comprising multiple organs.
Technological advances, however, allow the generation of increasingly
higher dimensional data, so we continue to progress towards a more
complete understanding of human disease. The next-generation sequencing
technologies are already having a major impact on DNA sequencing,
identifying rare variations in tumour tissues associated with different
cancer types. In addition, subsequent generations of sequencing
technologies are on the horizon and promise to deliver the sequence of
entire human genomes in days and at a reasonable cost. Sequencing
technologies can also be used to identify patterns of methylation, to
fully characterize the transcriptome and to identify transcripts that
are being actively translated.The advances of the sequencing revolution
therefore stand ready to provide unprecedented snapshots of complex
systems that will allow a more accurate network view, which in turn will
lead to models of disease that have greater predictive power.
The
primary aims of generating and mining large-scale biological data sets
are to learn the fundamental rules that govern complex living systems
and to derive, as a result, predictive models of their behaviour.
Without sophisticated mathematical algorithms capable of appropriately
integrating the large-scale data, and without high-performance computing
environments in which to apply these algorithms, it will be difficult to
build generally predictive models. Information-systems support services
will become increasingly critical both for building predictive models
and for representing complex states of knowledge and making such
knowledge accessible to researchers so that they may refine and correct
the models of disease. Recent successes in programming machines to mine
complex data to derive the fundamental laws of motion perhaps represent
a glimpse into the future of biology, in which machines may be able to
derive fundamental rules in complex living systems, given large-scale
data sets.
Gene networks
Networks generally provide a convenient framework for exploring the
context within which single genes operate. Networks are simply graphical
models that comprise nodes and edges and are convenient for visualizing
complex mathematical models that describe how variables of a system
associate with one another in different contexts of interest. For gene
networks associated with biological systems, the nodes in the network
typically represent genes, gene products or other important molecular
entities, and an edge between any two nodes indicates a relationship
between the corresponding genes, gene products or other molecular
entities.
Cells comprise many tens of thousands of proteins, metabolites, RNAs and
DNAs, all interacting in complex ways. In turn, complex biological
systems comprise many types of cells operating within and between the
many types of tissue that make up different organ systems, all of which
interact in complex ways to give rise to a vast array of phenotypes that
manifest themselves in living systems. Modelling the extent of such
relationships between molecular entities, between cells, and between
organ systems is a daunting task. Networks are a convenient framework in
which to represent the relationships among these different variables. In
the context of biological systems, a network can be viewed as a
graphical model that represents relationships among DNAs, RNAs, proteins,
metabolites and higher-order phenotypes such as disease state. In this
way, networks provide a way to visualize extremely large-scale, complex
relationships among molecular and higher-order phenotypes in any given
context
Biological networks comprise nodes, which represent molecular entities
that are observed to vary in the population under study (for example DNA
variations, RNA levels, protein states or metabolite levels). Edges
between the nodes represent relationships between the molecular entities,
and these edges can either be directed, indicating a cause–effect
relationship, or undirected, indicating an association or interaction.
For example, a DNA node in the network representing a given locus that
varies in a population of interest may be connected to a
transcript-abundance trait, indicating that changes at the particular
DNA locus induce changes in the levels of the transcript. The
potentially millions of such relationships represented in a network
define the overall connectivity structure, or topology, of the network.
The more classic pathway view represents molecular processes on an
individual level, whereas networks represent global (population-level)
metrics describing variations between individuals in a population of
interest; these variations in turn define the coherent biological
processes in the tissue or cells associated with the network. |