Table 1. Three unique features inherent in the DNA molecule.
|
Ability of DNA molecule to separate into separate strands |
|
Ability of DNA strands to reanneal or hybridize |
|
The inherent negative charge of phosphorous |
|
MOLECULAR & CELLULAR
NEUROBIOLOGY
Master Course Cognitive Neuroscience - Radboud
University, Nijmegen
|
|
|
|
Chapter 5: Molecular biological research methodology |
| Bioinformatics - data analysis | CRISPR-cas genome editing |
| ChIP-chip/seq |
|
|
Techniques used in Molecular Biology
|
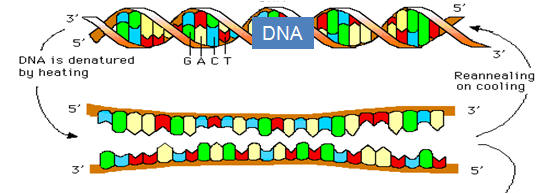
Three features inherent in the DNA molecule provide the foundation for research and medical applications Three features essential to all techniques of recombinant technology are worthy of note. The first is the ability of DNA, a double-stranded molecule, to denature and anneal or hybridize (Table 1). The double-stranded DNA, held together by hydrogen bonding of the corresponding complementary basis, will under exposure to high temperatures (95°C) separate into two strands and, if the temperature is reduced, the complementary strands will again come together (anneal) and return to their previous double-stranded state. The process of separating DNA into separate strands is referred to as denaturation and the recombining process is known as annealing or hybridization with the latter term preferred if the two DNA fragments are from different sources. It is notable that the two strands of DNA join together identically to that of the parent molecule. This is because of complementary base pairing, whereby A must bind with T and C to G. The third feature utilized in the various techniques is that of the high energy phosphorus, which is present in the DNA molecule, providing it with a negative charge. These properties are essential to all techniques as well diagnostic and therapeutic agents that arise or are derived from recombinant techniques. One exploits the property of DNA to denature and recombine whereby a complementary DNA marker can be labeled with either a radionuclide or a color pigment and, if the complementary DNA is present in the sample, it will recombine (hybridize) with the indicator molecule to confirm the presence of the complementary base pair. The probe is usually an oligonucleotide of 20 or 30 bases, which is adequate to isolate DNA molecules. One example of this is confirming the diagnosis of a pathogen. The negative charge imparted by the phosphorus is exploited to separate DNA molecules of different sizes through electrophoresis. By selecting the size of the pores in a media, one can separate molecules of different sizes based on their charge as they move toward the positive electrode. All techniques utilized in molecular biology utilize two or more of these procedures. |
|
|
Table 1. Three unique features inherent in the DNA molecule.
|
|
See also under "Molecular biology and Recombinant DNA technology".
|
Isolation of DNA DNA can be isolated from all human tissues with the exception of mature erythrocytes, which no longer contain a nucleus. Most procedures used in molecular biology require only nanogram (10−9 g) quantities of DNA, so enough tissue to yield these quantities may be obtained even from blood smears. In man, lymphocytes are a convenient and accessible source of DNA, since they have the added advantage that they can be transfected by a virus (usually Epstein–Barr virus) to produce an immortal cell line that can be propagated in the laboratory indefinitely under appropriate cell-culture conditions. This process is called lymphocyte transformation and provides for a continuous renewable source of DNA from an original specimen. However, today the technique of whole-genome amplification has all but replaced development of cell lines as a means of having renewable DNA. Regardless of the source, the DNA present within the nucleus must be extracted and separated from the other cellular components. This is performed by lysis of the cell and nuclear membrane, removal of all proteins, and isolation of the DNA. Classically, this procedure involves lysis of the cell using a lytic enzyme and a nonionic detergent, deproteinization of the lysed products using the organic solvents phenol and chloroform, and precipitation of the DNA by ethanol from a solution of high salt concentration. If performed appropriately, this procedure yields a fairly pure product visible to the naked eye. This process is universally used and has led to the production of automated instruments that effectively and efficiently extract the DNA such that 10 to 15 ml of whole blood will typically yield approximately 50 to 100 μg of genomic DNA.
|
|
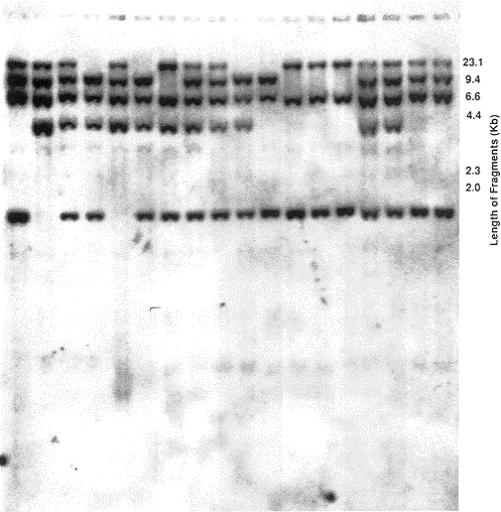
Separation of DNA fragments by gel electrophoresis After DNA is isolated, it is digested with a restriction endonuclease and loaded into a well of agarose or polyacrylamide gel and subjected to an electric current (Figure1). Each individual nucleotide because of its phosphorus has a net negative charge that forms the basis of separation of DNA fragments in an electrical current. The DNA fragments migrate toward the anode according to their size with the larger fragments migrating slowest. Following separation of the fragments, the gel is stained with ethidium bromide. To determine the size of the fragments of the unknown DNA, standard DNA fragments of known size are concomitantly electrophoresed for comparison. Electrophoresis through agarose will separate double-stranded DNA fragments varying from 1000 to 100,000 bp and polyacrylamide is used to separate DNA fragments from 6 to 1000 bp. Theoretically, polyacrylamide gels should separate fragments differing in size by only 1 bp. Utilizing these gels, one can detect bands as little as 1 ng and a difference between fragments of 0.5% of their size. |
|
|
|
FIG 1. A typical Southern blot with distinct bands. Each vertical lane consists of DNA from separate individual. |
|
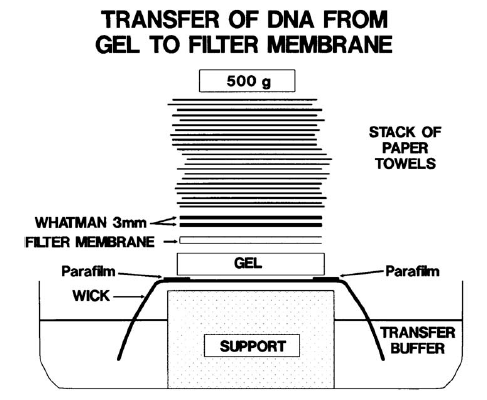
Southern, Northern, and Western blotting (see also under Detection of DNA, RNA and protein) The ability to identify specific fragments of DNA after electrophoresis became routine after the development of the Southern transfer technique (Figure 2) by E. M. Southern in 1975, now referred to as Southern blotting. He developed a procedure whereby DNA fragments separated by gel electrophoresis are transferred by capillary action to a filter and the pattern of the bands are permanently fixed identical to the pattern obtained on the gel. Briefly, the electrophoresed double-stranded DNA is chemically denatured into single strands while in the gel and passively transferred onto a nitrocellulose filter or a nylon membrane. The transferred DNA is then irreversibly bound to the filter by baking the filter at a high temperature. DNA can be effectively transferred by this procedure after several hours with the resultant blot being the exact duplicate of the gel containing the electrophoresed DNA. |
|
|
|
FIG 2. Southern transfer apparatus. |
|
The blotted membrane can then be used to identify particular segments of DNA by reacting (hybridizing) the single-stranded DNA on the membrane with a solution containing a probe such as a 32P radioactively labelled single-stranded segment of DNA, which is complementary to a DNA region within the targeted segment. The conditions of the hybridization process are empirically established such that the probe attaches only to it complementary segment of DNA, and the resultant double-stranded product is analogous to the native double-stranded DNA. The probe may be a segment of DNA defined by its sequence (oligonucleotide) or merely a small fragment of DNA from a much larger segment of interest. X-ray film is then exposed to the tagged membrane at −70°C in a process called autoradiography. The resultant pattern shown on the X-ray film reflects complementary fragments of DNA on the membrane that hybridized to the probe. Analysis of RNA by a similar technique is termed Northern blotting and analysis of proteins is termed Western blotting. In human disease caused by large DNA deletions rather than single-base pair changes, Southern blotting is the method of choice for detection. The use of a DNA probe covering the known sequence of the deletion will not be visible after hybridization of the probe if the deletion is present in the DNA segment, but will be visualized in a normal DNA fragment.
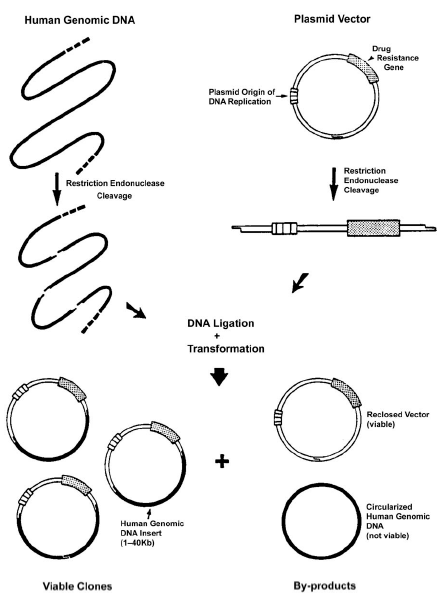
DNA cloning The prerequisites for cloning are depicted in Figure 3 and consist of the following: (1) an isolated DNA fragment to serve as an insert; (2) a vector (plasmid); (3) a restriction endonuclease site common to both the insert and the vector so that the DNA ends will be compatible, allowing the fragment to be ligated or inserted into the vector; (4) a DNA ligase to ligate the insert into the vector; and (5) a means to differentiate the host cells that have incorporated the vector with the insert from those that have not. |
|
|
|
FIG 3. The cloning of a DNA fragment utilizing a plasmid as the vector. |
|
Fundamentally, DNA cloning is a technique that replicates a specific fragment of DNA in a replicating organism. The foreign DNA fragment (insert) is ligated (inserted) into a larger segment of DNA (vector). The vector (containing insert) is then placed in its host cell, where it replicates and amplifies. There are several means of detecting those bacteria which have incorporated the vector (with insert). One approach is to modify the vector to include a gene that expresses a protein resistant to ampicillin. Another is to express a gene which emits a color. The bacteria are grown in media containing ampicillin and only those that have the resistance gene (and therefore the vector) will survive. Thus after the bacteria are grown and plated out on agar, monoclonal bacterial colonies (clones) are selected on the basis of ampicillin resistance. Growth in this media indicates that the vector was successfully incorporated into the bacterial colony and the vector with insert was successfully cloned. A single colony of bacteria is selected and further amplified in culture media. DNA fragments from any source can be amplified at least a million-fold. Following amplification, the vector containing the insert of interest is purified, and the cloned insert isolated in large quantities. Typically, vectors used are plasmids, circular DNA molecules that naturally replicate in various strains of bacteria. Inserts may represent any DNA fragment of interest, typically a gene coding for a protein of interest that may then be made in large quantities for biochemical studies. The route yield from cloning is about 1 million copies of the desired DNA fragment. Cloning is used by industry to produce for pharmaceutical purposes vast amounts of purified substances that normally are found in limited quantities. For instance, endocrinologists treat patients with diabetes mellitus using recombinant human insulin. See also under Molecular biology and Recombinant DNA technology.
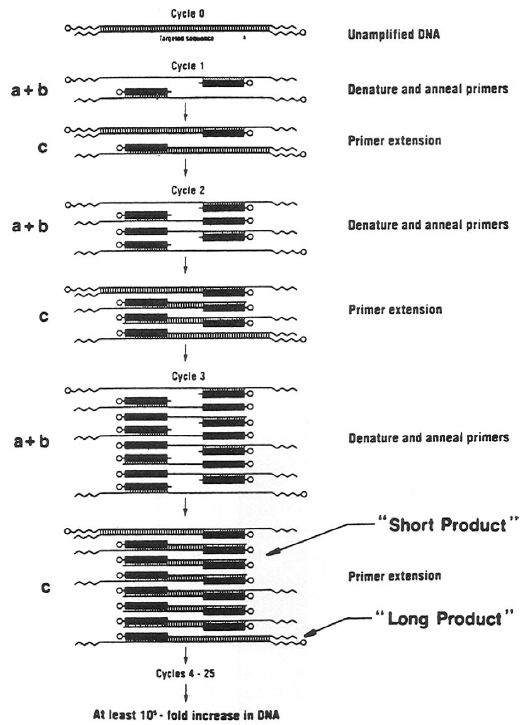
Polymerase Chain Reaction (PCR) The development of the PCR (Figure 4) revolutionized the approach to molecular biology and molecular genetics. PCR is an automated in vitro repetitive reaction that uses a heat-stable DNA polymerase to amplify a specified segment of DNA. The elegance of PCR lies in its simplicity and the property to amplify a single copy of the target fragment of DNA up to 1 million-fold in a matter of hours without the use of tedious classic cloning techniques, thus enabling easy access to large quantities of DNA. An essential property of DNA making PCR possible is the property of DNA to denature and separate into its two complementary strands and, under appropriate conditions, reanneal and again become double-stranded (hybridization). The complementary base pairing and the ability to hybridize underlies all the essential techniques of molecular genetics. |
|
|
|
FIG 4. Polymerase chain reaction (PCR).
|
|
Practically the only limitation to PCR is the need to know a short stretch of base sequences flanking both the 5′ and the 3′ ends of the desired segment of DNA. A sequence of approximately 20 bp on both ends of the desired fragment is all that is necessary. These oligonucleotide sequences, called primers, are complementary to small regions of both strands of target DNA (template) located at both ends (5′ and the 3′ end). A sense primer is designed with a sequence identical to the 5′ end of the template and an antisense primer is designed with the sequence of the strand that is the complement to the 3′ end of the template. Under the usual reaction parameters used, primers of 18 to 24 bp have a significant affinity for only the target template and, therefore, the vast majority of the reaction product is specific. The strategic location of the primers and the 5′ to 3′ action of DNA polymerase dictate that the amplified region will be everything located between the primers. A reaction cycle consists of denaturation of a single double-stranded DNA template; annealing of the primers to the specified regions on the template; and extension from both primer sites by DNA polymerase in the 5′ to 3′ direction, to yield two double-stranded products. This reaction is cyclically repeated to increase the number of products exponentially until a plateau is reached after approximately 25 to 30 cycles. It is routinely possible to amplify fragments up to 10,000 bp (10 kb). The applications of PCR are many. The PCR product, which may represent a portion of a gene, may be screened for a genetic mutation by one of numerous mutation detection systems. In principle, mutation detection systems are designed to detect a change in DNA sequence by observing shifts in gel mobility or melting properties of a mutant DNA fragment, as compared to a fragment with normal sequence. Mutation detection systems are sensitive to the level of detecting a single basepair alteration. A commonly used technique in human genetics is single-stranded conformation polymorphism (SSCP) analysis, whereby a single basepair change from normal sequence may show a differing migration pattern on a non-denaturing gel, caused by a change in secondary structure of the mutated fragment. Similarly, a mutated DNA sequence may alter the melting temperature of DNA, resulting in differential retention of the fragment (compared to normal sequence) on a chromatography column (denaturing high-performance liquid chromatography, DHPLC). Alternatively, a researcher may choose to directly sequence the nucleotide DNA sequence and compare the sequence to normal sequence. The process of PCR may also be used to derive full-length cDNA (gene sequence without intervening introns) from single-copy mRNA of expressed genes by a method called rapid amplification of cDNA ends. This method uses multiple primer pairs located within the region of interest as well as at the end of the mRNA sequence, including the poly(A) tail. Amplification using multiple primer sets generates multiple overlapping products; the overlapping regions can be identified and the remaining sequences can be combined to define the entire cDNA. The diagnostic utility of PCR has been well established in infectious disease, including viral myocarditis. Conventionally, detection of viral DNA required at least 50,000 copies of viral nucleic acid per cell, making the diagnosis of viral myocarditis virtually impossible using myocardial biopsy samples. Presently, single-copy viral genes can be amplified for detection by PCR. These are just a few of the potential applications of PCR and it is most likely that PCR will continue to have its impact on medicine for years to come. See also "PCR" under Molecular biological research methodology.
|
|
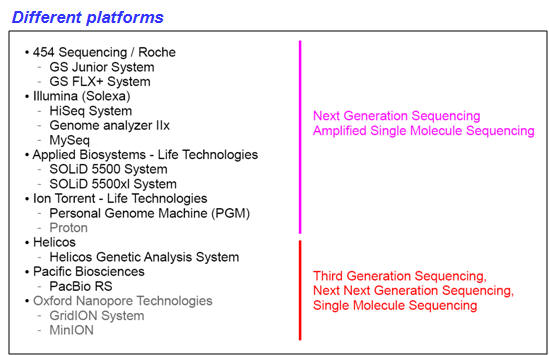
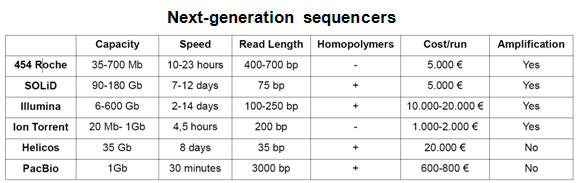
DNA sequencing - Next Generation Sequencing (NGS)
DNA sequencing is the determination of the precise sequence of nucleotides in a sample of DNA. The classical and until recently most commonly used method is called the dideoxy (Sanger) method. DNA sequencing reactions all use a primer to initiate DNA synthesis. This primer will determine the starting point of the sequence being read, and the direction of the sequencing reaction. The success of this technique is based on the use of fluorescently labeled dideoxy nucleotides for each of the bases comprising DNA (ddATP, ddCTP, ddGTP, ddTTP). As opposed to deoxynucleotides, dideoxynucleotides (ddNTPs) lack of a free 3′ OH group. Although ddNTPs may be added to a DNA template being elongated by DNA polymerase, these ddNTPs prevent the next nucleotide from being added, and the chain would terminate. In a sequencing PCR reaction, both normal and dideoxynucleotides are present and the incorporation of a ddNTP will occur randomly. Through repeated cycles of denaturing, primer annealing, and extension/termination, new strands of DNA may be synthesized that will all vary in where in the fragment termination occurred due to the incorporation of a fluorescently labeled ddNTP. Thus, at the conclusion of a DNA sequencing reaction, a pool of DNA exists ranging from the smallest possible fragment size of 1 bp to the maximize size of the fragment. In automated DNA sequencers, the different fluorescent labels attached to each of the four dideoxynucleotides (ddA, ddC, ddG, and ddT) will be detected from base 1 to the final base, providing a color-coded DNA sequence. DNA sequencing has become common place and new technologies have enhanced the rate and precision for sequence determination. It is presently possible to sequence many millions of base pairs per day using automated technology; the newest sequencing approach involves nanopore technologies (see tables and figure below). |
|
|
Applications: genome sequencing (e.g., de novo sequencing; resequencing; targeted (re)sequencing; mitochondrial sequencing; mutation detection); transcript expression profiling (e.g., RNA sequencing; miRNA sequencing; deep-SAGE; deep-CAGE); transcription factor binding (e.g., ChIP sequencing); structural variation; metagenomics. |
|
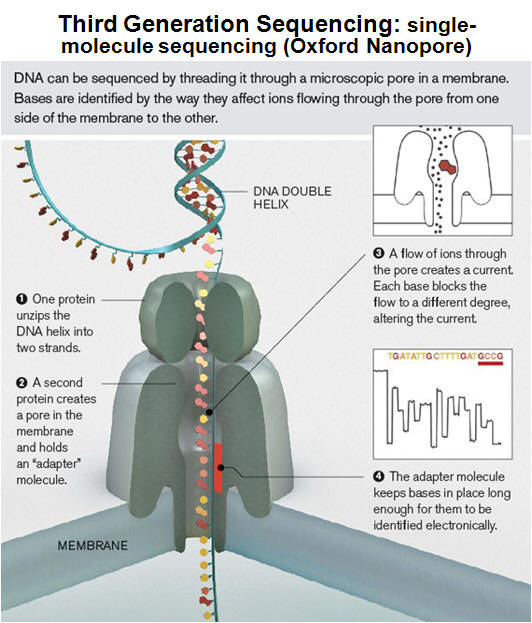 For a movie providing an overview of how nanopores can be used to sequence DNA, click here (or see: http://www.nanoporetech.com/news/movies#movie-24-nanopore-dna-sequencing) |
|
Site-directed mutagenesis The technique of site-directed mutagenesis provided researchers the ultimate tool for studying the pathophysiological basis of disease caused by genetic mutations. This technique was made possible by previous innovations in molecular biology, including PCR, insertion of a DNA fragment into a vector, and cloning. A circular plasmid molecule containing a gene of interest may serve as a template for PCR. Primers are designed to purposely mismatch a single nucleotide in the gene, engineering an altered codon, replacing one amino acid for another. Following PCR, the amplified plasmids will then contain the insert with the mutated codon, and the mutated gene may be cloned for further studies. The advances in molecular biology leading to the identification of disease-causing DNA mutations required a technique to generate mutant proteins and to analyze their function in vitro to elucidate the abnormal physiology leading to disease. Site-directed mutagenesis enables the study of mutant proteins in in vitro systems and recombinant DNA molecules can also be injected into the germ line of mice and successfully expressed in succeeding generations. These transgenic animals thereafter become a model for in vivo analysis of the pathogenesis of the disease.
|
|
|
| Next page: Genetic transmission | Go back to: Molecular biology and Recombinant DNA technology |
|
|