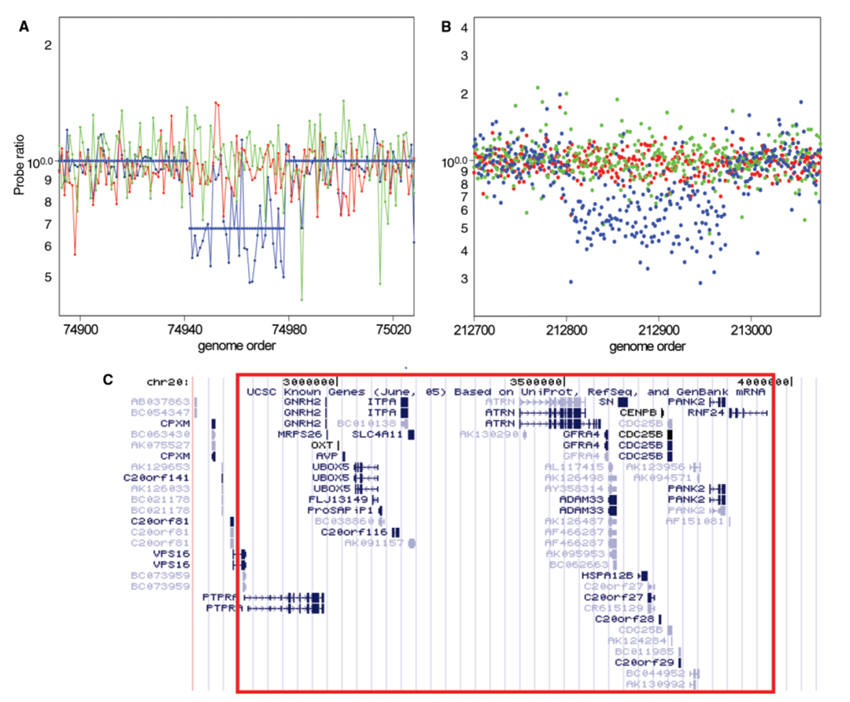
As we saw in the previous sections, a major challenge in Neuroscience is to understand the genetic basis of higher brain functions in human. One strategy is to take advantage of the ever-growing knowledge about the nucleotide sequences of whole genomes and to search for a link between genetic variations in these genomes and differences in brain functions. Single-Nucleotide Polymorphisms (SNPs) are single-nucleotide differences (polymorphisms) or one-letter variations in the DNA sequence that occur when a single nucleotide (A, T, C, or G) in the genome sequence is altered. Recently, it became clear that also genomic rearrangements resulting in copy number variations (CNVs) represent important genetic variations among individuals.
Single-Nucleotide Polymorphism (SNP)
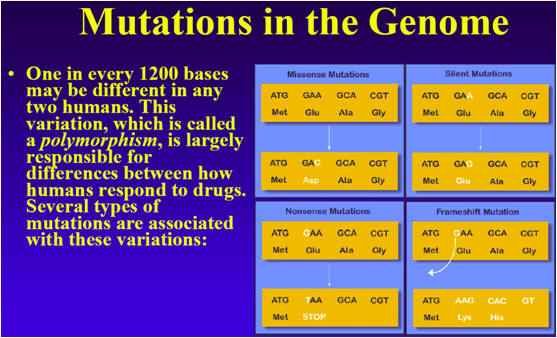

SNPs are caused when nucleotides replicate less than perfectly or mutate. This replication process is enormously complex, and happens perfectly most of the time, but occasional imperfections or mutations do occur. In humans, the mutations occur at about 200 mutations per generation. Thus, each child receives about 200 such mutations from his father, and 200 from his mother, for a total of 400 mutations, spread more-or-less randomly across the genome. Because each child will only pass on about half of the father's mutations to the next generation, virtually all of the father's 200 mutations will be gone after 20 generations or 400 years. Occasionally, however, one persists and becomes established in the population.
There are millions of SNPs in the human genome. In fact, it is estimated that if any two genomes are compared, there will be on average one SNP every 1,200 base pairs. SNPs thus contribute to differences among individuals that are present in humans with a frequency of about once in every 1,200 bases. Most of the mutations have no effect on the way an individual looks or functions. A small fraction of mutations are beneficial, allowing the species to evolve and adapt to new environments, while others are quite catastrophic. Yet other mutations produce only subtle effects, not noticeable by themselves, but sufficient to produce a larger effect when combined with other mutations.
The causes of common disease are very complex. We know that both the environment and genetics play important roles. Research in human genetics has revealed that the majority of common human diseases, as well as most human responses to medicines, involve the interaction of many genes i.e. they are polygenic. This makes understanding the underlying causes extremely difficult, as they are likely to be influenced by a large number of SNPs found in a variety of genes and intergenic regions that act together in specific combinations. By itself, each individual SNP may have only a small effect on disease susceptibility or drug response.
This complexity highlights the futility of attempting to identify disease causality (or drug response) through analysis of individual genes, small regions of chromosomes, or even whole chromosomes. Instead, detailed analysis of entire genomes is required to identify these polygenic effects. The challenge is to be able to identify which of the millions of SNPs throughout the entire genome contribute to a particular disease or drug response.
A high-density oligonucleotide array approach offers the ability to analyze large numbers of SNPs in large numbers of individuals using automated methods. The arrays consist of DNA probes (short segments of DNA) that are synthesized in pre-determined positions on glass surfaces. Sample DNA hybridized to the arrays will bind in such a way that the genotype of each of the SNPs can be determined. The probes are designed with multiple redundancy, thus increasing the accuracy with which each SNP is assayed. Methods have been developed to use such high-density oligonucleotide arrays to genotype more than 1.5 million SNPs in either pooled or individual DNA samples in an accurate, rapid, and cost-effective manner.

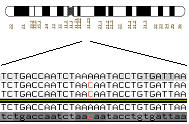
Finding sequence variation: examine multiple sequences from the same genomic region.
Microarray-based SNP-detection
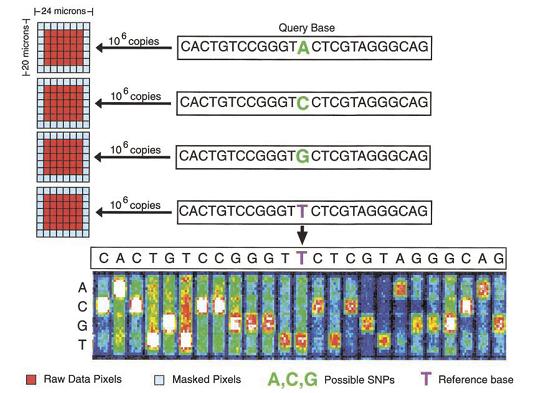
High-throughput SNP detection using microarrays. Eight features (four for the forward strand and four for the reverse complement strand) are associated with every queried site. Each feature consists of a 25-base oligonucleotide. The 13th base is the query base and all possible genotypes are tested