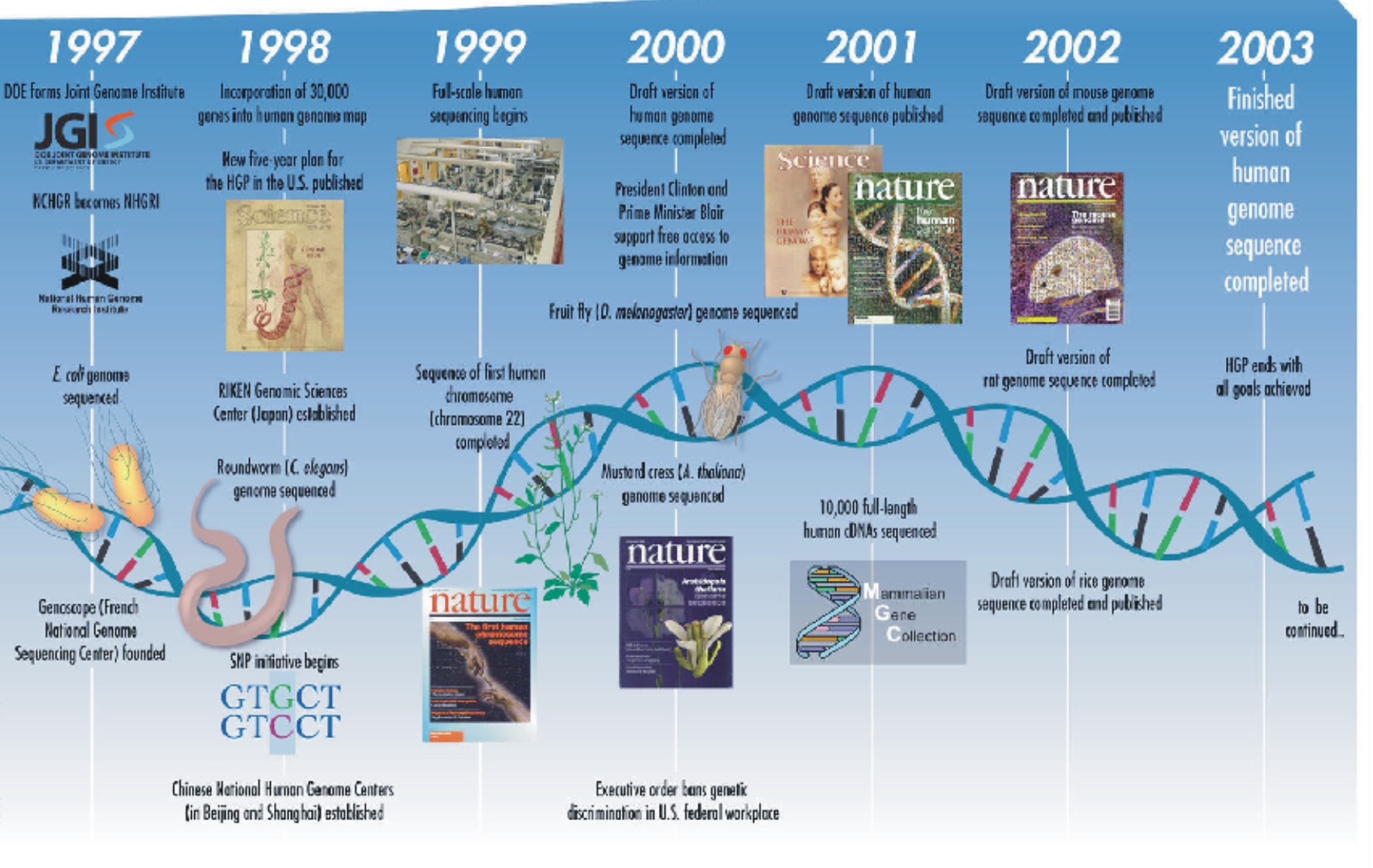
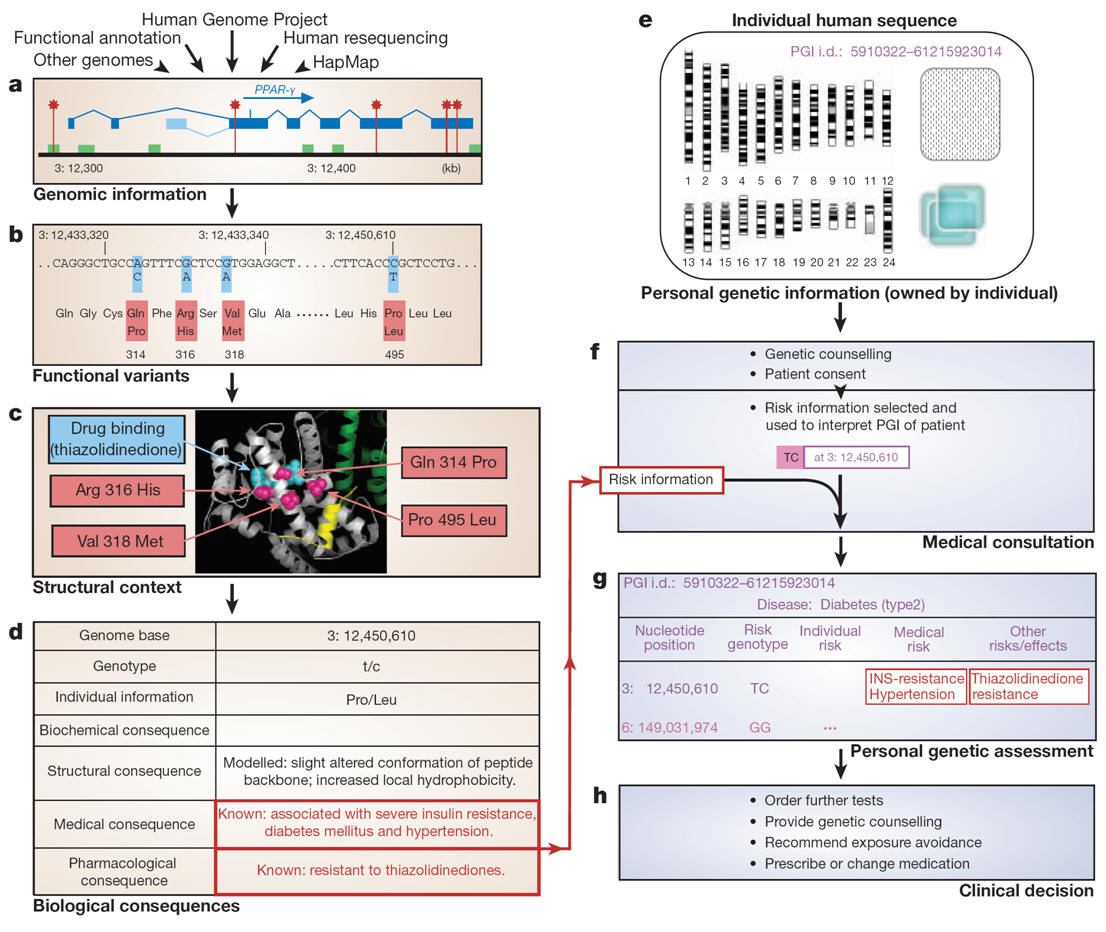
-
The human genome contains 3164.7 million chemical nucleotide bases (A, C, T, and G).
-
The average gene consists of 30,000 bases, but sizes vary greatly, with the largest known human gene being dystrophin at 2.4 million bases.
-
The total number of genes is estimated at 21,000 — much lower than previous estimates of 80,000 to 140,000 that had been based on extrapolations from gene-rich areas as opposed to a composite of gene-rich and gene-poor areas.
-
Almost all (99.9%) nucleotide bases are exactly the same in all people.
-
The functions are unknown for over 50% of discovered genes.
-
Less than 2% of the genome codes for proteins.
-
Repeated sequences that do not code for proteins ("junk DNA") make up at least 50% of the human genome.
-
Repetitive sequences are thought to have no direct functions, but they shed light on chromosome structure and dynamics. Over time, these repeats reshape the genome by rearranging it, creating entirely new genes, and modifying and reshuffling existing genes.
-
During the past 50 million years, a dramatic decrease seems to have occurred in the rate of accumulation of repeats in the human genome.
How it is arranged
-
The human genome's gene-dense "urban centers" are predominantly composed of the DNA building blocks G and C.
-
In contrast, the gene-poor "deserts" are rich in the DNA building blocks A and T. GC- and AT-rich regions usually can be seen through a microscope as light and dark bands on chromosomes.
-
Genes appear to be concentrated in random areas along the genome, with vast expanses of noncoding DNA between.
-
Stretches of up to 30,000 C and G bases repeating over and over often occur adjacent to gene-rich areas, forming a barrier between the genes and the "junk DNA." These CpG islands are believed to help regulate gene activity.
-
Chromosome 1 has the most genes (2968), and the Y chromosome has the fewest (231).
How the human compares with other organisms
-
Unlike the human's seemingly random distribution of gene-rich areas, many other organisms' genomes are more uniform, with genes evenly spaced throughout.
-
Humans have on average three times as many kinds of proteins as the fly or worm because of mRNA transcript "alternative splicing" and chemical modifications to the proteins. This process can yield different protein products from the same gene.
-
Humans share most of the same protein families with worms, flies, and plants, but the number of gene family members has expanded in humans, especially in proteins involved in development and immunity.
-
The human genome has a much greater portion (50%) of repeat sequences than the mustard weed (11%), the worm (7%), and the fly (3%).
-
Although humans appear to have stopped accumulating repeated DNA over 50 million years ago, there seems to be no such decline in rodents. This may account for some of the fundamental differences between hominids and rodents, although gene estimates are similar in these species. Scientists have proposed many theories to explain evolutionary contrasts between humans and other organisms, including those of life span, litter sizes, inbreeding, and genetic drift.
Click Chimp & humans diverge from a common ancestor for an animation.
Click
Comparison human &
Neanderthaler
for an animation.
Variations and mutations
-
There are about 1.4 million locations where single-base DNA differences (single-nucleotide polymorphisms, SNPs) occur in humans. This information promises to revolutionize the processes of finding chromosomal locations for disease-associated sequences and tracing human history.
-
The ratio of germline (sperm or egg cell) mutations is 2:1 in males vs females. Researchers point to several reasons for the higher mutation rate in the male germline, including the greater number of cell divisions required for sperm formation than for eggs.
Applications, future challenges
Deriving meaningful knowledge from the DNA sequence will define research through the coming decades to inform our understanding of biological systems. This enormous task will require the expertise and creativity of tens of thousands of scientists from varied disciplines in both the public and private sectors worldwide. The draft sequence already is having an impact on finding genes associated with disease. A number of genes have been pinpointed and associated with breast cancer, muscle disease, deafness, and blindness. Additionally, finding the DNA sequences underlying such common diseases as cardiovascular disease, diabetes, arthritis, and cancers is being aided by the human variation maps (SNPs) generated in the Human Genome Project in cooperation with the private sector. These genes and SNPs provide focused targets for the development of effective new therapies. One of the greatest impacts of having the sequence may well be in enabling an entirely new approach to biological research. In the past, researchers studied one or a few genes at a time. With whole-genome sequences and new high-throughput technologies, they can approach questions systematically and on a grand scale. They can study all the genes in a genome, for example, or all the transcripts in a particular tissue or organ or tumor, or how tens of thousands of genes and proteins work together in interconnected networks to orchestrate the chemistry of life.
Anticipated benefits
-
improved diagnosis of disease
-
earlier detection of genetic predispositions to disease
-
rational drug design
-
gene therapy and control systems for drugs
-
personalized, custom drugs